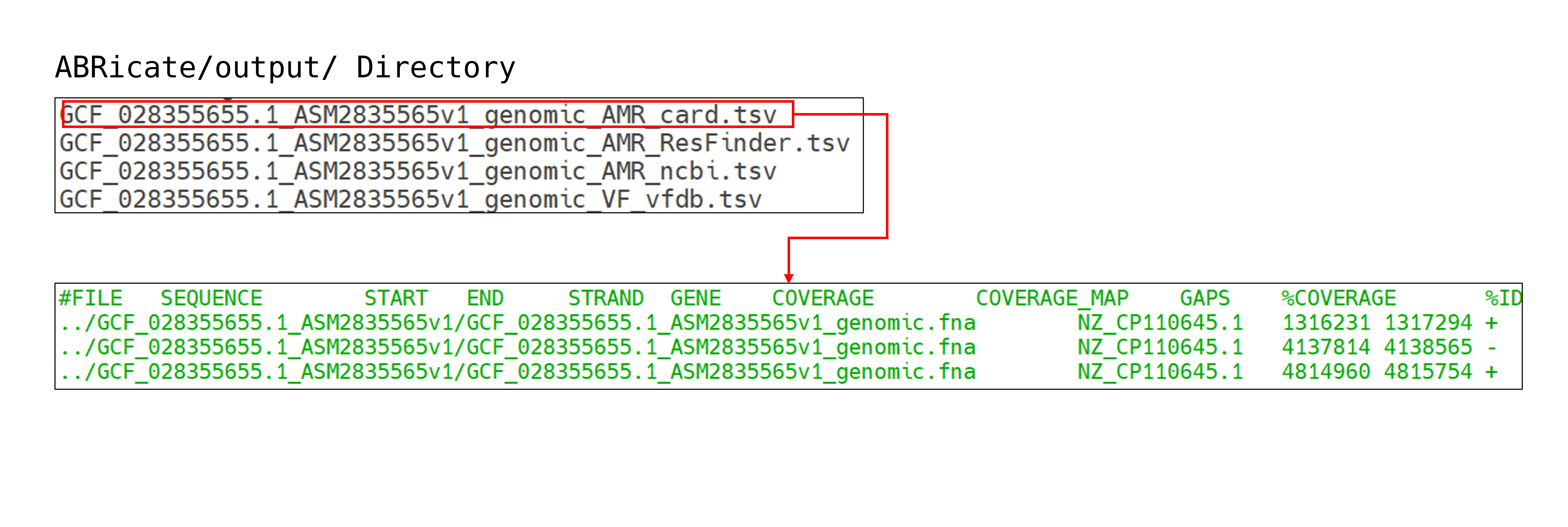
박테리아의 유전체 어셈블리(genome assembly)가 완료된 후에도, 해당 샘플의 정확한 분류학적 위치를 파악하거나 항생제 내성 및 병원성 유전자의 존재 여부를 확인하기 위해서는 따로 후속 분석이 필요합니다. 이 파이프라인은 GTDB-Tk, MLST, ABRicate의 세 가지 도구를 사용해 이러한 기능을 제공하고 있습니다. 세 프로그램 모두 어셈블리가 완료된 유전체 서열 정보를 FASTA 형식의 파일로 입력받아 분석을 시작합니다. 종 동정(Species Identification) 분석은 GTDB-Tk를 사용하여 수행됩니다. 이 도구는 입력된 유전체 서열을 GTDB에 존재하는 방대한 박테리아 서열 정보와 비교 분석하여, 해당 샘플에 대한 종 수준의 분류학적 정보(Taxonomy)를 출력합니다. 이후 MLST 도구를 사용해 종 수준보다 하위 분류군인 균주(Strain) 정보를 확인할 수 있습니다. 해당 도구는 유전체 전체 서열이 아닌, 하우스키핑 유전자(house-keeping gene) 서열만을 활용하여 분류를 수행합니다. 마지막으로 ABRicate를 사용하여 박테리아 유전체 서열 내에 존재하는 항생제 내성 유전자 및 병원성 유전자에 대한 정보를 확인합니다. 이를 위해 CARD, ResFinder, NCBI AMRFinderPlus, VFDB 등 다양한 전문 데이터베이스의 서열 정보를 이용할 수 있습니다. 이러한 단계별 분석을 통해 단순한 서열 정로부터 taxonomy, 임상적 특성을 포괄적으로 파악할 수 있습니다.
- 카테고리 Genomics > Bacterial Pathogen Analysis
- 수정일2025-11-07
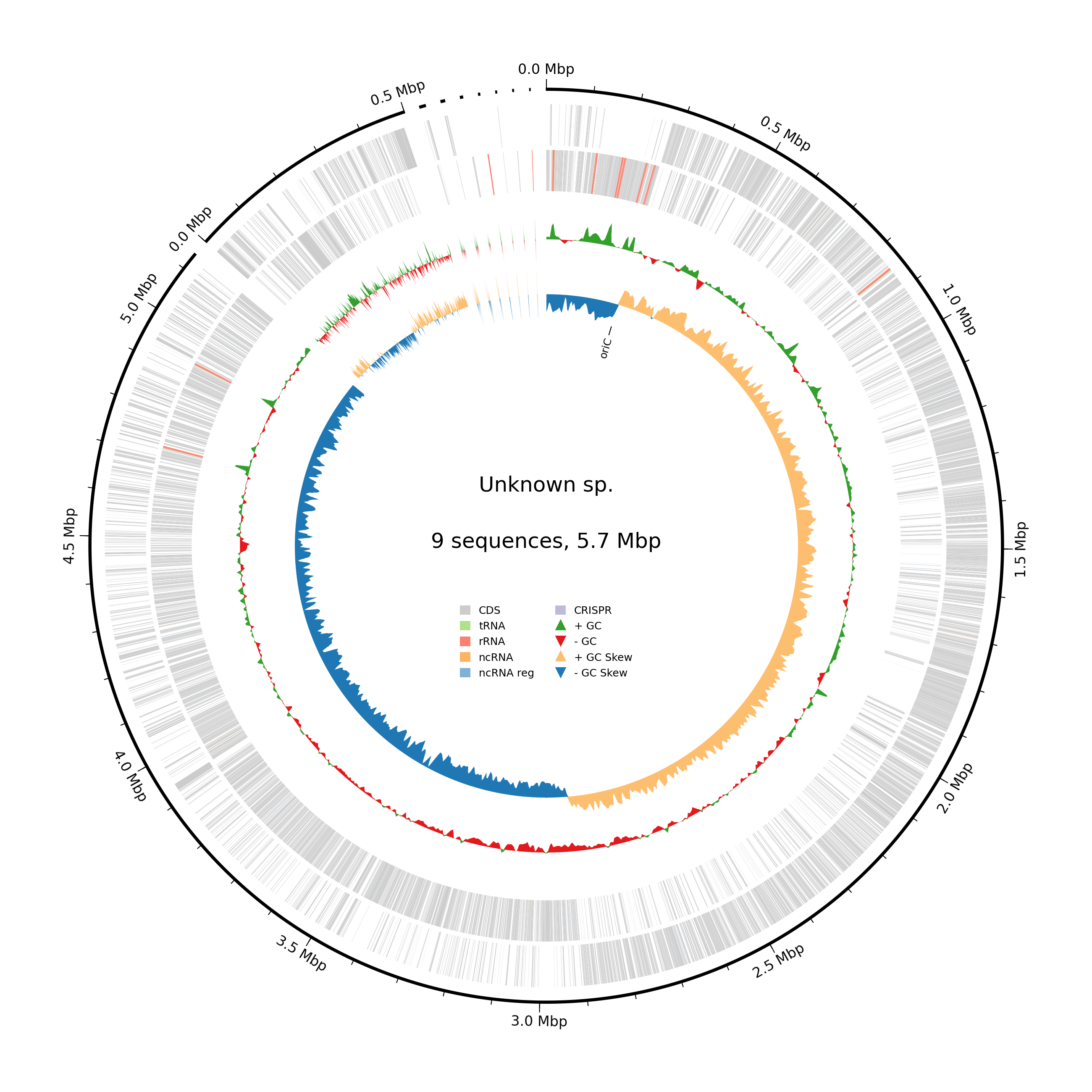
Bacterial Genome Assembly Pipeline은 bacterial WGS 데이터를 기반으로 QC부터 annotation까지 원스텝으로 분석을 수행할 수 있는 자동화 파이프라인입니다. 해당 Pipeline은 ZGA라는 프로그램을 기반으로 수행되며, read QC, read processing, de novo assembly, genome polishing, assembly QC, annotation 단계로 구성되어 있습니다. 또한, 분석자가 시각점과 끝나는 단계를 지정해서 분석을 수행할 수 있는 장점도 존재합니다. 파이프라인의 초기 단계(Read QC, read processing)에서는 실험 데이터의 품질 평가와 정제가 이루어집니다. fastp를 사용하여 실험 데이터의 품질을 검사하고 평가하며, BBtools의 BBDuk을 활용하여 시퀀싱 어댑터 및 낮은 품질의 리드를 효과적으로 정제합니다. 또한, BBtools의 BBMerge 단계에서 각 paired-reads를 미리 overlap하여 assembly의 효율을 증가시킵니다. 마지막으로 사용자의 선택에 따라 Mash를 사용하여 예상되는 genome size를 예측할 수 있습니다. 두 번째 단계(de novo assembly, genome polishing)에서는 Unicycler, SPAdes, Flye라는 3개의 assembly tool 중에서 하나를 선정하여 de novo assembly를 수행합니다. 그 후, 각 assembly tool에 내장된 기능을 바탕으로 assembled sequence를 polishing하여 genome의 품질을 향상시킵니다. 이어지는 단계(Assembly QC, annotation)에서는 checkM을 이용하여 genome completeness, contamination, heterogeneity를 확인하여 assembled genome의 품질을 확인합니다. 마지막으로 Bakta를 이용하여 assembled genome에 대한 annotation을 수행하는 것으로 Bacterial Genome Assembly Pipeline은 종료됩니다. 전체적으로, 최상위 입력 데이터인 fastq 형식의 bacteria WGS raw data로부터 시작하여 QC, de novo assembly, assembly QC, annotation까지 분석하실 수 있습니다.
- 카테고리 Genomics > Large-genome-assembly
- 수정일2025-11-10
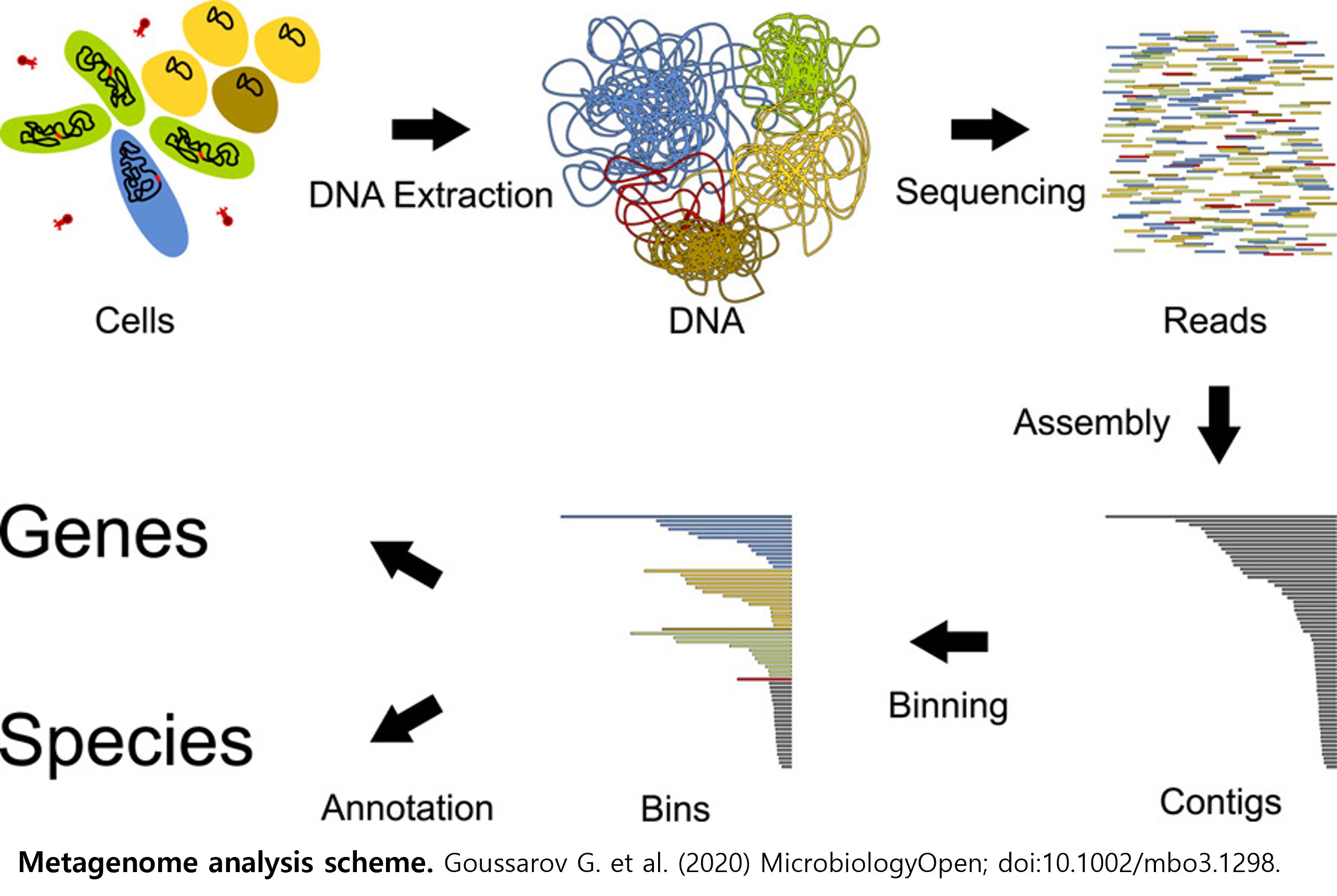
MAG(Metagenome-Assembled Genome) 분석 파이프라인은 복합 미생물 군집의 시퀀싱 데이터를 이용하여 배양되지 않은 미생물의 유전체를 복원하고, 이를 계통학적 및 기능적으로 분석하기 위한 통합 분석 절차입니다. 이 파이프라인은 원시 시퀀싱 데이터로부터 품질 관리(QC), 어셈블리(assembly), binning, 품질 평가(QC of bins), 계통 분류(taxonomic classification) 및 기능 주석(annotation) 단계로 구성됩니다. 우선 품질 과정 단계를 통해 시퀀싱 데이터의 품질을 확인하고 저품질 리드 및 숙주 서열을 필터링하여 제거합니다. 이후 MEGAHIT를 이용해 정제된 리드로부터 contig를 조립하고, metaQUAST로 어셈블리 품질을 평가합니다. 어셈블리된 contig는 MetaBAT2, MaxBin2, CONCOCT를 이용해 유사한 염기 조성과 커버리지 패턴을 기준으로 유전체 단위(bin)으로 분류됩니다. 그 후 DAS Tool을 이용하여 각 도구의 binning 결과를 통합함으로써 중복을 제거하고 완전도와 정확도를 향상시킨 MAG 세트를 확보합니다. 생성된 MAG에 대해서는 CheckM2를 통해 완전도와 오염도를 평가하고 GTDB-Tk로 계통 분류를 수행합니다. 마지막으로 Prokka를 이용해 예측된 유전자에 대한 기능 주석을 수행합니다. 본 파이프라인은 시료별(single-sample)로 독립 수행되며, MAG 확보와 기능적 해석을 위한 절차로 활용됩니다.
- 카테고리 Metagenomics > Whole-genome-based
- 수정일2025-11-10
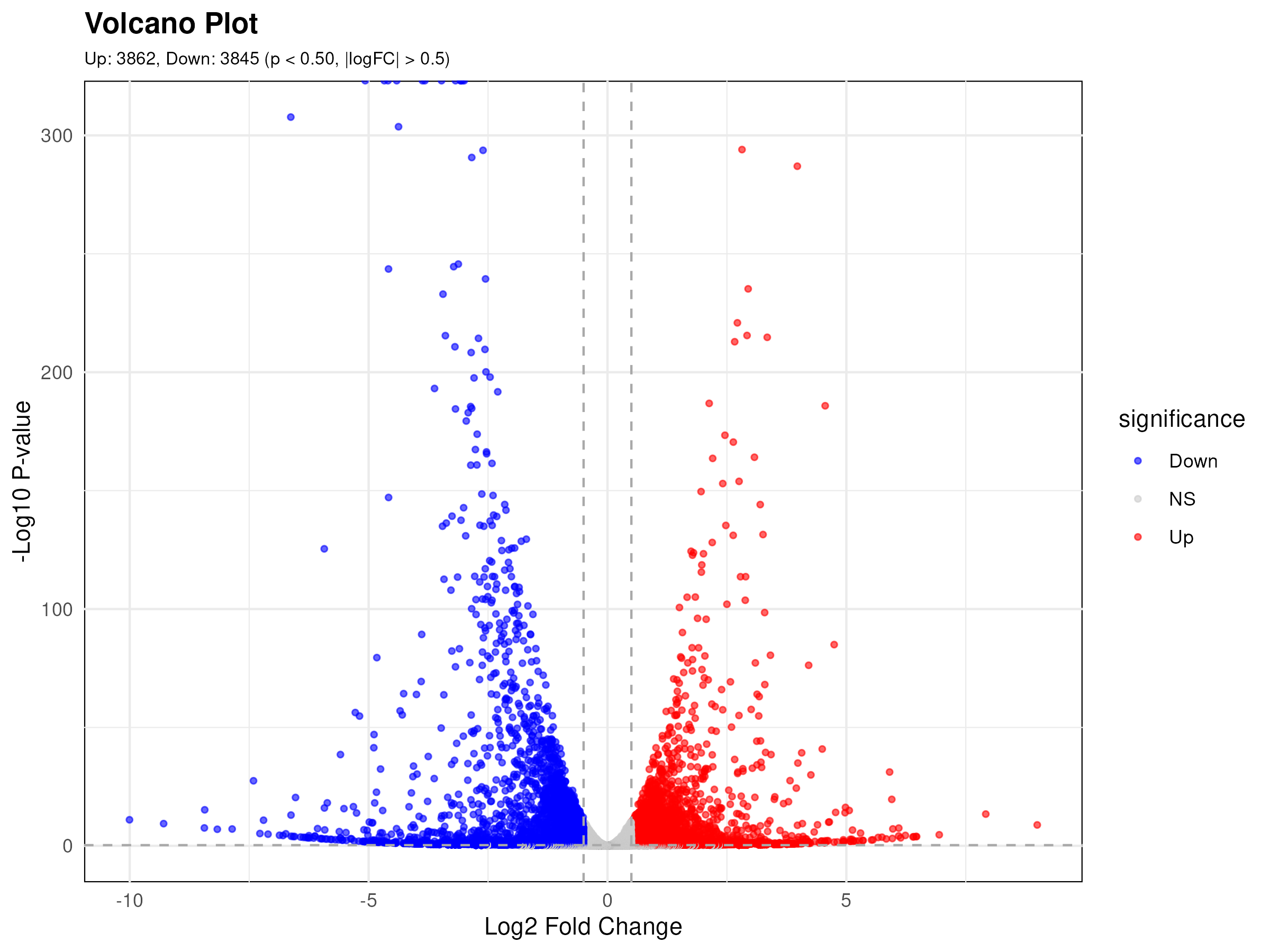
RNA-seq Analysis Pipeline은 RNA-Seq 데이터를 처리하고 유전자 발현에 대한 통계적 분석을 수행하는 것을 목표로 합니다. 이 파이프라인은 유전자의 발현 수준을 이해하고 해석하기 위해 실험 데이터의 품질을 평가하고 정제하며, 정렬, 발현 수준 계산, 통계적 분석, 결과 시각화로 구성되어 있습니다.파이프라인의 초기 단계에서는 실험 데이터의 품질 평가와 정제가 이루어집니다. FastQC를 사용하여 실험 데이터의 품질을 검사하고 평가하며, Cutadapt를 활용하여 시퀀싱 어댑터 및 낮은 품질의 리드를 효과적으로 정제합니다.다음으로, STAR 2를 이용하여 리드를 정확하게 유전체에 정렬하고, 각 유전자의 발현 위치를 정밀하게 파악합니다. 그 후, Rsubread 라이브러리의 FeatureCounts를 활용하여 정렬된 리드를 각 유전자에 할당하여 발현 수준을 정량화하고 Count Matrix를 생성합니다.이어지는 단계에서는 R 기반의 edgeR과 limma를 사용하여 발현 수준의 통계적 차이를 식별하고 각 유전자의 발현 변동을 분석합니다. 이 과정에서 control과 test 샘플에는 각각 최소 두 개 이상의 생물학적 복제 샘플이 포함되어야 통계적 분석이 가능하다는 점에 유의해야 합니다. 복제가 없는 경우 잔차 유도가 0이 되어 분석이 실패하거나 결과가 신뢰성을 잃을 수 있습니다. 또한, R 기반의 fgsea를 활용하여 gene set 간의 풍부도를 평가하고, 다양한 시각화 도구를 활용하여 효과적으로 표현합니다. 마지막으로, fgsea의 결과 파일을 이용하여 여러 R 패키지를 통해 데이터 시각화, 그래픽 생성 등 실험 결과를 자세히 분석하고 시각화합니다.전체적으로, 최상위 입력 데이터인 fastq 형식의 RNA-seq raw data로부터 시작하여 품질 보고서인 fastqc.report.html을 생성하고, MA plot, correlation plot, network, volcano plot, heatmap 등의 다양한 시각화 자료를 통해 유전자 발현 및 풍부도를 시각적으로 확인할 수 있습니다. Bio-Express RNA-seq Alternative-splicing Pipeline (이하 AS)은 유전자 발현의 전사체 수준에서 크게 5가지 type의 splicing 양상을 확인할 수 있다.Alternative splicing은 하나의 유전자를 구성하는 복수의 exon (coding region)간의 조합에 따라 여러 transcripts (isoforms)가 생성되며, 이에 따라 하나의 유전자라도 서로 다른 구조를 갖는 단백질이 만들어짐에 따라 기능이 다른 유전자로써 역할을 하게된다. 이러한 메커니즘을 통해 단백질의 폭넓은 capacity 를 확보할 수 있으며, 다양한 분자적 역할이 가능하다. (출처: From Wikipedia, the free encyclopedia) 기본적인 5가지 형태의 이벤트는 아래와 같다. 1. SE (Exon skipping cassette exon) 2. MXE (Mutually exclusive exons) 3. A5SS (Alternative donor site) 4. A3SS (Alternative acceptor site) 5. RI (Intron retention) AS 분석 파이프라인은 아래와 같은 흐름으로 진행됨. 1. Quality Control, 시퀀싱 품질 관리 (by FastQC) 2. Trimming, 아답터 및 low quality 제거 (by Cutadapt, Trimmomatic) 3. Mapping, 레퍼런스 alignment (by STAR, HISAT2) 4. AS detection, 선택적 스플라이싱 탐색 (by rMATs) 5. Visualization, AS 결과 시각화 (by ggsashimi)
- 카테고리 Transcriptomics > RNA-seq-transcriptomics
- 수정일2025-11-07
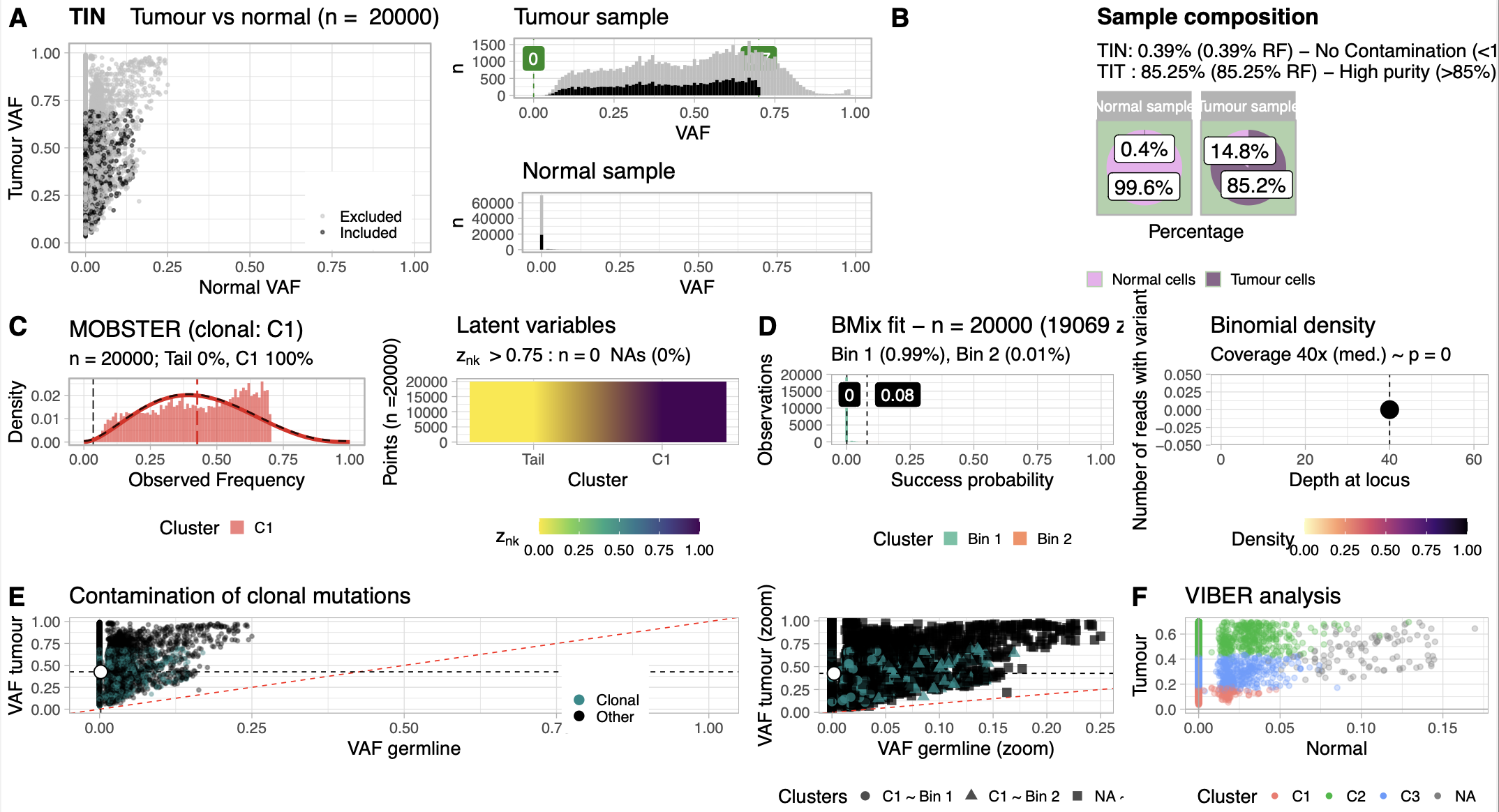
Bio-Express Somatic WGS Pipeline은 전장 유전체 시퀀싱 데이터로부터 체세포 변이를 검출하기 위한 모듈식 분석 파이프라인입니다. 이 파이프라인은 raw FASTQ 파일을 입력 데이터로 사용하고, 종양-정상 쌍 분석을 기반으로 하는 포괄적인 체세포 변이 호출 결과와 품질 평가 및 시각화를 제공합니다. FastQC를 통한 시퀀싱 품질 평가 후, Cutadapt로 어댑터 제거 및 품질 트리밍을 수행하고, BWA-MEM2 정렬 도구를 사용하여 참조 유전체 서열에 매핑하여 BAM 형식의 정렬 파일을 생성합니다. 이후 GATK 파이프라인을 통해 중복 제거, 매핑 품질 평가, 그리고 저품질 read에 대한 필터링을 수행하며 모든 페어 정보가 일치하는지 확인합니다. SAMtools를 활용한 좌표 기준 정렬과 GATK MarkDuplicates를 통한 PCR 중복 제거를 거쳐, GATK BaseRecalibrator와 ApplyBQSR을 사용하여 알려진 변이 사이트 정보를 공변량으로 활용한 염기 품질 점수 재보정을 수행합니다. 재보정이 완료된 BAM 파일에 대해 먼저 포괄적인 품질 관리 및 샘플 검증 단계를 수행합니다. Somalier를 통한 샘플 관계 검증, SNPmatch를 활용한 변이-SNP마커 통합 분석을 통한 샘플 정체성 확인, VerifyBamID2를 통한 샘플 오염도 평가, 그리고 Mosdepth를 사용한 커버리지 분석을 통해 시퀀싱 데이터의 품질과 신뢰성을 종합적으로 평가합니다. 이어서 종양-정상 쌍 분석 단계로 진입하며, Conpair를 통한 Normal-Tumor 페어 적합성 검증과 교차 개체 오염 수준 추정을 수행합니다. 그 다음 Strelka2와 Mutect2를 통한 단일 염기 변이 및 삽입/결손 변이 검출을 병행하여 체세포 변이의 민감도와 특이도를 극대화합니다. 마지막으로 TINC를 통한 종양 순도 분석과 Manta를 사용한 구조 변이 호출, Canvas를 이용한 복제수 변이 분석으로 포괄적인 체세포 유전체 변화를 정량화하여 암 유전체학 연구와 정밀 의학에 필수적인 정보를 제공합니다. > 기본 참조 게놈: hg38 [중요] 샘플 유형 식별 방법: - 종양 조직 샘플: FASTQ 파일명에 "_T" 포함 필수 - 정상 조직 샘플: FASTQ 파일명에 "_N" 포함 필수 (예시) patient001_T_R1.fastq.gz # 종양 샘플, Read 1 patient001_T_R2.fastq.gz # 종양 샘플, Read 2 patient001_N_R1.fastq.gz # 정상 샘플, Read 1 patient001_N_R2.fastq.gz # 정상 샘플, Read 2
- 카테고리 Genomics > Variant-analysis
- 수정일2025-10-30
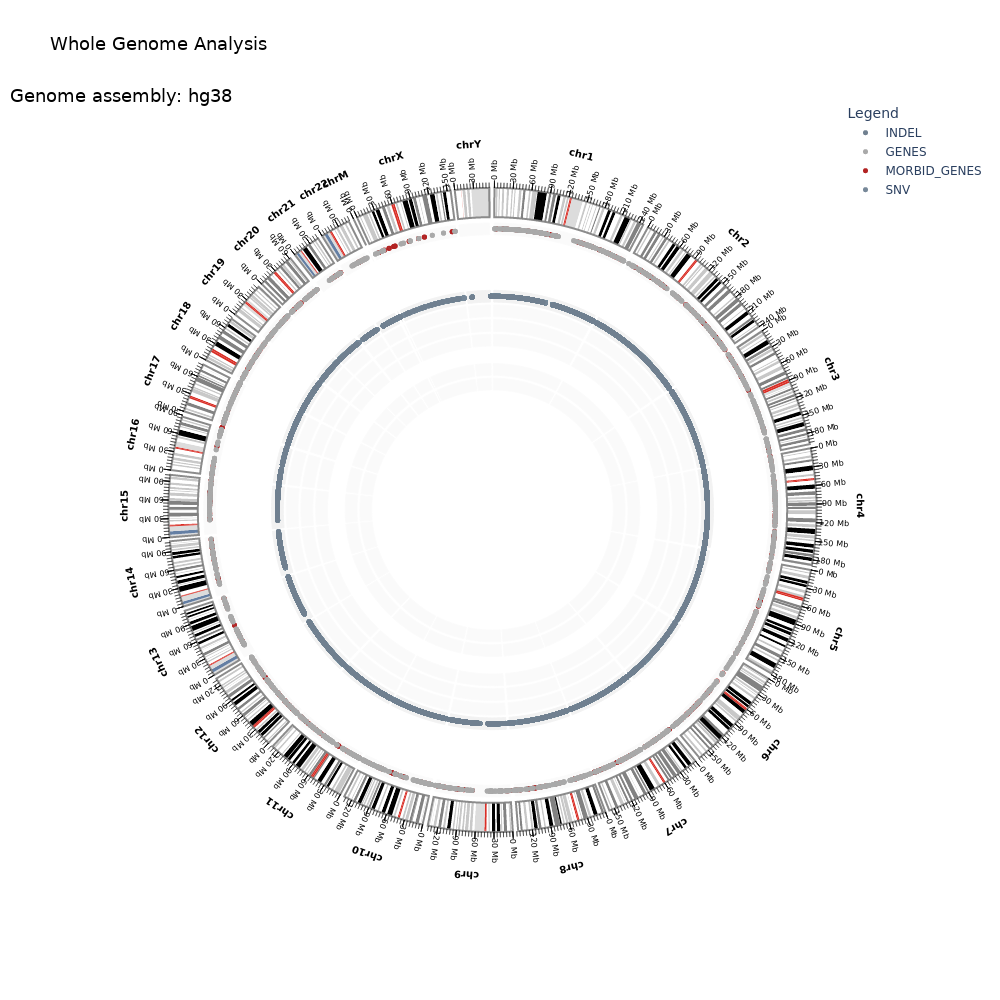
Bio-Express Germline WGS Pipeline은 전장 유전체 시퀀싱 데이터로부터 생식세포 변이를 검출하기 위한 모듈식 분석 파이프라인입니다. 이 파이프라인은 raw FASTQ 파일을 입력으로 사용하고, 개체 유전체 분석을 기반으로 허눈 포괄적인 생식세포 변이 호출 결과와 품질 평가 및 시각화를 제공합니다. FastQC를 통한 시퀀싱 품질 평가 후, Cutadapt로 어댑터 제거 및 품질 트리밍을 수행하고, BWA-MEM2 정렬 도구를 사용하여 참조 유전체 서열에 매핑하여 BAM 형식의 정렬 파일을 생성합니다. 이후 GATK 파이프라인을 통해 중복 제거, 매핑 품질 평가, 그리고 저품질 read 필터링을 수행하며 모든 페어 정보가 일치하는지 확인합니다. SAMtools를 활용한 좌표 기준 정렬과 GATK MarkDuplicates를 통한 PCR 중복 제거를 거쳐, GATK BaseRecalibrator와 ApplyBQSR을 사용하여 알려진 변이 사이트 정보를 공변량으로 활용한 염기 품질 점수 재보정을 수행합니다. 재보정이 완료된 BAM 파일에 대해 먼저 포괄적인 품질 관리 및 샘플 검증 단계를 수행합니다. Somalier를 통한 샘플 관계 검증, VerifyBamID2를 통한 샘플 오염도 평가, 그리고 Mosdepth를 사용한 커버리지 분석을 통해 시퀀싱 데이터의 품질과 신뢰성을 종합적으로 평가합니다. 이어서 GATK HaplotypeCaller를 이용한 GVCF 파일 생성 및 GenotypeGVCFs를 활용한 표준 VCF 형태의 생식세포 SNV/Indel 변이 탐지를 실행합니다. 후속적으로 BCFtools를 적용한 종합적인 변이 통계 해석을 진행하며, Manta 도구를 통해 구조적 변이를 검출합니다. > 기본 참조 게놈: hg38
- 카테고리 Genomics > Variant-analysis
- 수정일2025-10-30
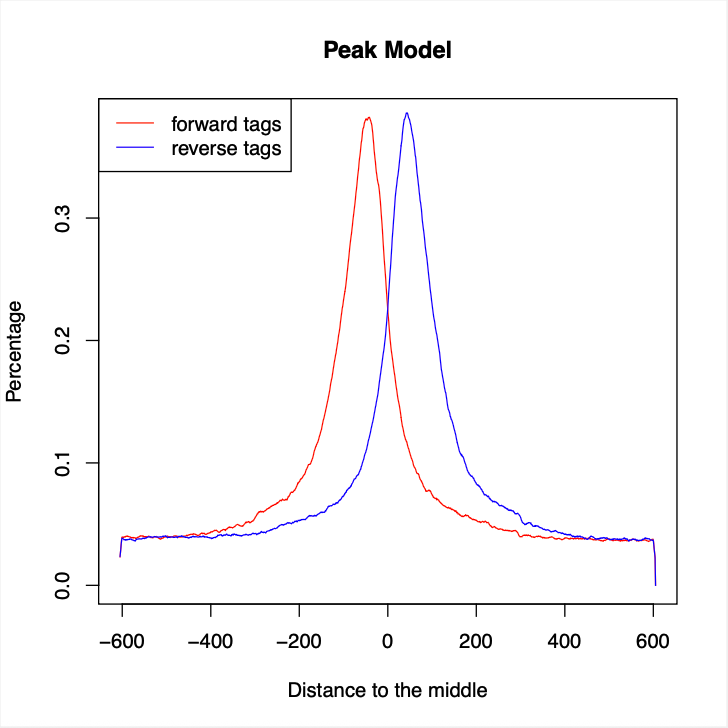
Bio-Express ChIP-seq Analysis Pipeline은 크로마틴 면역침전 시퀀싱(Chromatin Immunoprecipitation Sequencing) 데이터로부터 단백질-DNA 결합 부위를 검출하기 위한 모듈식 분석 파이프라인입니다. 이 파이프라인은 raw FASTQ 파일을 입력으로 사용하고, 전사인자 결합 사이트, 히스톤 변형 영역, 크로마틴 구조 분석을 기반으로 하는 포괄적인 후성유전학적 결합 부위 호출 결과와 품질 평가 및 시각화를 제공합니다. FastQC를 통한 시퀀싱 품질 평가 후, FASTX-Toolkit을 사용하여 저품질 염기 필터링을 진행하고, Bowtie2 정렬 도구를 사용하여 참조 유전체 서열에 매핑하여 SAM 형식의 정렬 파일을 생성합니다. 이후 전처리가 완료된 정렬 파일을 활용하여 후성유전학적 신호 분석 단계로 진입합니다. MACS2(Model-based Analysis of ChIP-Seq)를 통한 통계적으로 유의한 피크 호출을 수행하여 단백질-DNA 결합 부위를 정확히 식별하고, narrowPeak 형식으로 고해상도 결합 영역을 제공합니다. 최종적으로 Homer를 활용한 포괄적인 후속 분석 단계를 수행합니다. annotatePeaks 기능을 통해 검출된 피크의 게놈 위치 주석과 주변 유전자 정보를 제공하고, makeUCSCfile을 사용하여 UCSC 게놈 브라우저와 호환되는 bedGraph 형식의 시각화 파일을 생성하여 크로마틴 면역침전 신호의 게놈 전체 분포 패턴을 직관적으로 확인할 수 있습니다. > 기본 참조 게놈: hg38 [중요] 샘플 유형 식별 방법: - 컨트롤 파일: "CONTROL_"로 시작 필수 (자동 식별을 위한 필수 접두사) - 처리/ChIP 파일: 특별한 파일명 규칙 없음 (예시) CONTROL_input_R1.fastq.gz # 유효한 컨트롤, Read 1 CONTROL_input_R2.fastq.gz # 유효한 컨트롤, Read 2 ChIP_H3K4me3_R1.fastq.gz # 유효한 처리군, Read 1 ChIP_H3K4me3_R2.fastq.gz # 유효한 처리군, Read 2
- 카테고리 Epigenomics > DNA-binding-protein-based-analysis
- 수정일2025-10-31
.png)
Single-cell RNA sequencing pipeline은 10X Genomics의 Cell Ranger, python 패키지인 Scanpy를 이용하여 단일 세포 수준의 유전자 발현 데이터를 분석하기 위한 파이프라인입니다. 본 파이프라인은 raw sequencing data(FASTQ 파일)과 함께 각 library가 유래한 샘플의 정보를 함께 입력받습니다. 이후 count matrix 생성, data preprocessing, normalize and scaling, dimension reduction and clustering, annotation, differential gene expression analysis 및 functional analysis 등을 수행합니다. 결과물로 cell-by-gene expression matrix 및 이로부터 파생된 분석 결과 테이블, 시각화한 그래프 등을 얻을 수 있습니다.
- 카테고리 Transcriptomics > Single-cell-transcriptomics
- 수정일2025-09-23