- 카테고리 Metagenomics > Whole-genome-based
- 수정일2025-11-10 10:05:28
- 레퍼런스
MAG(Metagenome-Assembled Genome) 분석 파이프라인은 복합 미생물 군집의 시퀀싱 데이터를 이용하여 배양되지 않은 미생물의 유전체를 복원하고, 이를 계통학적 및 기능적으로 분석하기 위한 통합 분석 절차입니다. 이 파이프라인은 원시 시퀀싱 데이터로부터 품질 관리(QC), 어셈블리(assembly), binning, 품질 평가(QC of bins), 계통 분류(taxonomic classification) 및 기능 주석(annotation) 단계로 구성됩니다.
우선 품질 과정 단계를 통해 시퀀싱 데이터의 품질을 확인하고 저품질 리드 및 숙주 서열을 필터링하여 제거합니다. 이후 MEGAHIT를 이용해 정제된 리드로부터 contig를 조립하고, metaQUAST로 어셈블리 품질을 평가합니다. 어셈블리된 contig는 MetaBAT2, MaxBin2, CONCOCT를 이용해 유사한 염기 조성과 커버리지 패턴을 기준으로 유전체 단위(bin)으로 분류됩니다. 그 후 DAS Tool을 이용하여 각 도구의 binning 결과를 통합함으로써 중복을 제거하고 완전도와 정확도를 향상시킨 MAG 세트를 확보합니다. 생성된 MAG에 대해서는 CheckM2를 통해 완전도와 오염도를 평가하고 GTDB-Tk로 계통 분류를 수행합니다. 마지막으로 Prokka를 이용해 예측된 유전자에 대한 기능 주석을 수행합니다.
본 파이프라인은 시료별(single-sample)로 독립 수행되며, MAG 확보와 기능적 해석을 위한 절차로 활용됩니다.
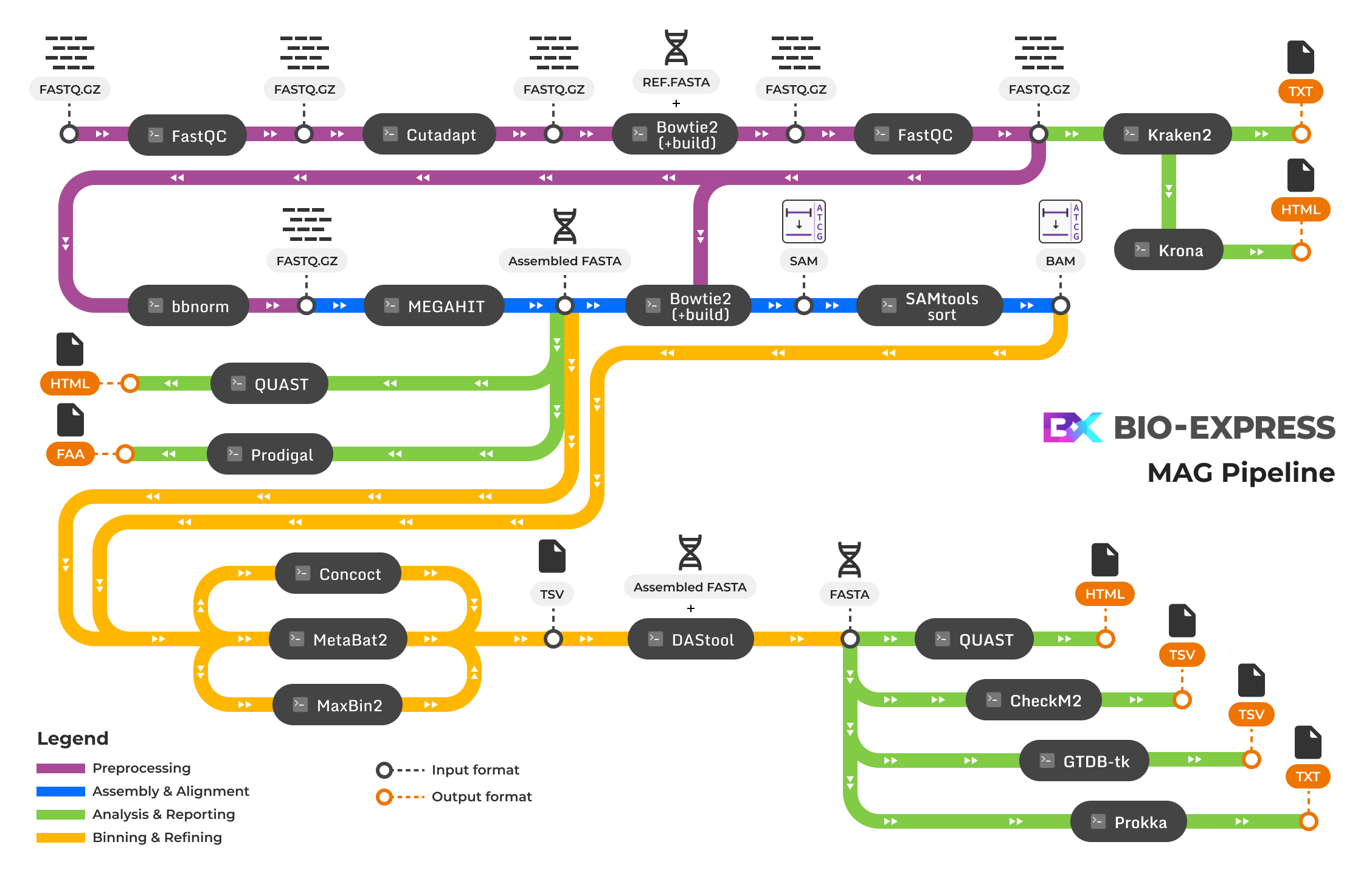
파이프라인 모듈
FastQC
FastQC는 고속 염기서열 분석(high throughput sequence) 데이터의 품질 관리를 위한 분석도구입니다. 이 프로그램은 FASTQ 형식의 서열 데이터를 읽어들여 여러 품질 관리(Qaulity Control) 검사를 수행하고 결과는 HTML 기반의 보고서로 출력합니다. FastQC는 전반적인 품질 문제에 대한 개요 정보를 제공하며, 쉽게 확인할 수 있는 요약된 그래프와 테이블을 포함합니다. FastQC는 FASTQ 형식의 파일이 입력 파일로 사용되며, 출력 결과는 리포트 html 파일과 zip 형식의 압축 파일이 생성됩니다.
주요사항
- FastQC는 자바 애플리케이션입니다. 실행하기 위해서는 시스템에 적절한 자바 실행 환경(Java Runtime Environment, JRE)이 설치되어 있어야 합니다. 따라서 FastQC를 실행하기 전에 먼저 적절한 JRE가 설치되어 있는지 확인해야 합니다. 다양한 종류의 JRE를 사용할 수 있지만, 저희가 테스트해본 것은 최신 오라클 런타임 환경과 adoptOpenJDK 프로젝트의 JRE입니다. 64비트 JRE를 다운로드하여 설치하고, 자바 애플리케이션이 시스템 경로(path)에 포함되도록 설정해야 합니다(대부분의 설치 프로그램이 이를 자동으로 처리해줍니다).
실행 명령어 예시
./fastqc.sh \ input_dir="./input" \ output_dir="./fastqc/output"
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./input | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | ./fastqc/output | 실행 후 결과물을 저장할 디렉토리 경로 (-o) |
결과
-
.png)
Basic Statistics 테이블은 주어진 FASTQ 파일에 대한 간단한 통계적 정보를 제공합니다. 일반적으로 다음과 같은 정보를 포함합니다. Filename : 분석된 파일의 이름 또는 경로 File type : FASTQ 파일의 종류 Encoding : 품질 점수 인코딩 방식 Total Sequence : 총 서열 수 Filtered Sequences : Read 품질이 좋지 않은 서열 수 Sequence length : 서열의 길이 %GC : 서열에서의 GC 백분율
-
.png)
X축은 리드의 염기 위치를 나타내며, Y축은 품질 점수를 의미합니다. 점수가 높을수록 품질이 좋습니다. 중앙의 빨간색 선은 중앙값을 나타내고, 노란색 박스는 사분위간 범위(25~75%)를 의미합니다. 위쪽 및 아래쪽의 위스커(whisker)는 각각 10% 및 90% 포인트를 나타냅니다. 파란색 선은 평균 품질을 의미합니다. 어떤 염기의 하위 사분위수가 10 미만이거나 중위값이 25보다 작을 경우 경고로 간주됩니다. 또한, 어떤 염기의 하위 사분위수가 5 미만이거나 중위값이 20보다 작으면 오류로 간주됩니다.
-
.png)
색을 이용하여 각 타일의 품질을 나타내며, 파란색은 품질이 높음을 나타내고 빨간색은 품질이 낮음을 나타냅니다. 각 타일의 품질을 모든 타일의 평균 품질과 비교하여 예상 패턴과의 편차를 식별 할 수 있습니다. 특정 타일의 품질이 지속적으로 좋지 않으면 물리적 결함이나 오염 등 셀의 특정 영역에 문제가 있음을 나타낼 수 있습니다. 이상적으로는 모든 타일이 높은 품질을 보여야 하면 플롯에서 더 차가운 색상으로 표시됩니다. 이 플롯은 Illumina 라이브러리에서만 나타납니다.
-
.png)
X축은 리드의 전체 길이에 대한 평균 품질 점수를 나타내고, Y축은 해당 품질 점수를 갖는 읽기의 수를 나타냅니다. 시퀀스의 시퀀싱 품질은 해당 시퀀스에 대한 정확성과 신뢰성을 나타내며, 품질 점수가 높을수록 오류 발생 가능성이 낮다는 것을 의미합니다. 만약 시퀀싱 실행이 전반적으로 낮은 품질을 보인다며, 시퀀싱 화학 문제나 샘플 준비 문제 등이 있을 수 있습니다. 품질 점수가 기록되지 않은 BAM/SAM 파일의 경우 확인할 수 없습니다. 품질 점수가 Phred 척도 기준 최고 품질 점수 27점(오류율 0.2%) 미만일 경우 경고 발생, 20점(오류율 1%) 미만일 경우 오류입니다.
-
.png)
X축은 리드의 포지션을 나타내고, Y축은 시퀀싱한 리드에서 각 base의 전체 비율을 나타냅니다. 좋은 품질의 시퀀싱 샘플에서는 각 위치의 염기 비율을 나타내는 4개의 선이 평행하고 서로 가까워야 합니다. 그러나 선이 일부 위치에서 엉키거나 얽히면 과도하게 표현된 시퀀스가 오염되었음을 나타낼 수 있습니다. 또한, A/T 또는 G/C 염기의 비율이 어떤 위치에서는 10% 이상 차이나면 경고 발생, 20%를 초과하면 오류입니다.
-
.png)
시퀀스에서 G와 C 뉴클레오티드의 백분율 비율을 나타냅니다. 이를 통해 DNA 또는 RNA 시퀀스의 특성을 이해할 수 있습니다. 시퀀스의 GC 함량은 DNA 안정성, 서열의 물리적 특성, 유전자 발현에 영향을 미칠 수 있으므로 중요한 지표 중 하나입니다. X축은 GC contents의 비율을 나타내고, Y축은 시퀀스의 총량을 나타냅니다. 정규 분포와 편차 합계가 전체 리드의 15%를 초과하면 경고, 30%를 초과하면 오류입니다.
-
.png)
시퀀싱 리드의 각 위치에서 발견된 N 비율을 나타내며 일반적으로 매우 낮습니다. 그러나 어떤 위치에서는 N 비율이 5%를 초과하면 시퀀싱 시스템에 문제가 있을 수 있다는 경고로 간주되며, 20%를 초과하면 오류로 간주됩니다. 데이터의 품질이 높은지 확인하고 시퀀싱 읽기의 정확성에 영향을 줄 수 있는 문제를 식별하려면 시퀀싱 중에 N 비율을 모니터링하는 것이 중요합니다. N 비율이 권장 임계값을 초과하는 경우 문제의 심층적인 분석이 필요할 수 있습니다.
-
.png)
시퀀싱 데이터에서 각 리드의 길이에 대한 분포를 나타냅니다. 이 그래프의 X 축은 시퀀스 길이를, Y 축은 리드 수를 나타냅니다. 시퀀싱 데이터에서 발견된 리드의 길이가 어떻게 분포되어 있는지를 시각적으로 확인할 수 있습니다. 이 그래프를 통해 시퀀싱 데이터 세트의 리드 길이 분포를 파악할 수 있으며, 예상치 못한 리드 길이나 이상한 분포를 감지하여 데이터의 품질을 평가하는 데 도움이 됩니다. 일반적으로 시퀀스 길이 분포는 일정하거나 특정한 패턴을 따르지만, 비정상적인 분포는 시퀀싱 데이터에 문제가 있을 수 있다는 신호일 수 있습니다.
-
.png)
중복된 시퀀스가 전체의 20% 이상일 경우 경고, 50% 이상일 경우 오류입니다.
-
.png)
시퀀싱 데이터에서 빈번하게 등장하는 시퀀스를 나타내는 테이블입니다. 이 테이블은 시퀀싱 데이터에서 특정 시퀀스가 기대보다 더 자주 나타나는 경우를 식별합니다. 실험과정에서 발생한 오류, PCR 이중성, 어댑터 오류 또는 샘플의 비정상적인 특정으로 인해 발생할 수 있습니다. 해당 내용을 확인하고 잠재적인 문제를 식별하는 것은 시퀀싱 데이터의 정확성과 신뢰성을 높이는 데 도움이 됩니다.
-
.png)
시퀀싱 데이터에서 발견된 어댑터 시퀀스의 누적 백분율을 보여주는 차트입니다. 시퀀스가 발견된 위치에 따라 백분율이 증가하며, 시퀀스가 리드의 끝까지 존재하는 동안 계산됩니다. 어댑터의 비율을 확인하여 데이터의 품질을 평가하며, 어댑터 시퀀스가 발견되는 비율이 높을수록 시퀀싱 데이터에 오류가 포함될 가능성이 높아지므로, 5%를 초과하면 경고, 10%를 초과하면 오류입니다.
Cutadapt
Cutadapt는 NGS 시퀀싱 데이터에서 어댑터(adapter) 서열 제거, 품질이 낮은 서열 제거 등 다양한 전처리 작업을 수행하는 데 사용되는 도구입니다. 주로 서열의 3’ 말단에서 염기서열을 제거하며, 특히 RNA 시퀀싱 또는 DNA 시퀀싱 데이터에서 어댑터 제거에 빈번하게 활용됩니다. 전체 데이터의 품질을 유지하여 이후의 분석 단계에서 정확한 결과를 얻을 수 있도록 도움을 줍니다. 입력 데이터로는 fastq, fastq.gz, fq, fq.gz 확장자를 가진 파일을 사용할 수 있습니다. 출력으로는 파일 확장자 앞쪽에 ‘_trimmed’라는 문구가 붙은 fastq 파일이 생성되도록 세팅되어 있습니다. 따라서 "trimmed.fastq" 파일은 프라이머 잔여물이나 다른 불순물이 제거된 reads 파일을 나타냅니다.
주요사항
- 1. Bio-Express의 Cutadapt 모듈은 paired-end와 single-end 형식의 FASTQ 데이터를 모두 처리할 수 있도록 설계되었습니다.
- 2. -a 및 –A 옵션에 입력해야 할 각각의 어댑터 서열은 NGS 시퀀서 플랫폼별로 다음과 같습니다. > Illumina: 기본값 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT > MGI: 필요시 기본값 대체 입력 -a AAGTCGGAGGCCAAGCGGTCTTAGGAAGACAA -A AAGTCGGATCGTAGCCATGTCGTTCTGTGAGCCAAGGAGTTG
실행 명령어 예시
$PROGRAM_DIR/cutadapt -q 20 –Q 20 -j 6 -a AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC -A AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 --pair-filter=$PAIR_FILTER -o $OUTPUT_DIR/$READ_1 –p $OUTPUT_DIR/$READ_2 $INPUT_DIR/$READ_1 $INPUT_DIR/$READ_2
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | path/to/input_dir | FASTQ 형식의 파일이 있는 디렉토리 경로 | |
| Output | Folder | output_dir | path/to/output_dir | 실행 후 결과물을 저장할 디렉토리 경로 (-o, -p) | |
| Option | Integer | min_len_r1 | 70 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read1의 최소 길이 | |
| Option | Integer | min_len_r2 | 1 | --minimum-length $MIN_LEN_R1:$MIN_LEN_R2 어댑터 제거 후 남아 있는 Read2의 최소 길이 | |
| Option | String | pair_filter | any | --pair-filter (single-end FASTQ에는 미적용) - any: 두 Read 중 어느 하나라도 조건에 부합하면 필터링 - both: 두 Read 모두 조건에 부합하면 필터링 | |
| Option | String | adapter_pos | adapter | 어댑터 시퀀스 처리 위치 지정 --adapter: 서열의 3' 끝 방향에서 어댑터를 찾아 해당 어댑터와 그 이후 모든 서열을 제거 (-a, -A) --front: 서열의 5' 시작 방향에서 어댑터를 찾아 해당 어댑터와 그 이전 모든 서열을 제거 (-g, -G) --anywhere: 5' 또는 3' 어느 쪽에든 나타날 수 있는 어댑터를 탐지 (-b, -B) | |
| Option | String | adapter_r1 | AGATCGGAAGAGCACACGTCTGAACTCCAGTCAC | 첫 번째 Read의 어댑터 시퀀스 (-a) | |
| Option | String | adapter_r2 | AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGT | 두 번째 Read의 어댑터 시퀀스 (-A) | |
| Option | Integer | quality_r1 | 20 | 첫 번째 Read의 절단 기준으로 사용할 품질 임계값 (-q) | |
| Option | Integer | quality_r2 | 20 | 두 번째 Read의 절단 기준으로 사용할 품질 임계값 (-Q) |
결과
-
.png)
총 입력된 read pair 수, 어댑터가 검출된 read의 수와 비율, 필터링 후 최종적으로 출력된 read 수 등 전체 리드 처리 현황을 요약함. 또한 데이터 전처리 단계에서 몇 %의 리드가 어댑터에 의해 잘렸고, 몇 %가 최종 분석에 사용 가능한지, 데이터 손실이 얼마나 발생했는지 파악함.
-
.png)
어댑터 서열 (Illumina adapter 등), 탐지 및 제거된 횟수, 최소 overlap 길이, mismatch 허용 개수 (error tolerance), 제거된 어댑터 서열에서의 염기 비율(어댑터 클리핑의 정확성 평가에 활용) 등 Read1에서 탐지된 어댑터의 정보. Read2에 대한 내용도 동일한 방식으로 출력함.
kraken2
Kraken2은 시퀀싱 데이터 전처리 과정을 통해 정제된 리드 데이터를 이용하여, 샘플 내에 존재하는 미생물의 구성(taxonomic composition)을 신속하게 파악하기 위한 분석 단계입니다. Kraken2는 K-mer 기반의 초고속 분류 알고리즘을 사용하여 각 리드가 어떤 생물 종 또는 상위 분류군에 속하는지를 식별합니다. 이때, 미리 구축된 참조 데이터베이스(reference database)와 입력 리드의 K-mer 정보를 비교하여 가장 높은 일치도를 갖는 계통 분류(taxonomic classification) 결과를 산출합니다.
이 과정은 어셈블리 이전에 수행되어, 샘플 별 미생물 군집의 대략적인 구성을 미리 파악하거나 오염 여부를 검증하는 품질 관리(QC) 용도로 활용됩니다.
실행 명령어 예시
kraken2.sh input_dir=./bowtie2-unmapped/output output_dir=./kraken2/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./bowtie2-unmapped/output | 분석에 사용될 fastq 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./kraken2/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 | |
| Option | Float | confidence | 0.0 | 분류를 허용할 최소 신뢰도(threshold score)를 설정, 0 ~ 1 사이의 값 지정 가능. Default: 0.0 | |
| Option | Integer | minimum-base-quality | 0 | 분류 시 고려할 최소 염기 품질(Phred 점수)을 설정. Default: 0 | |
| Option | Integer | minimum-hit-groups | 2 | 하나의 분류 호출(call)을 위해 요구되는 최소 “히트 그룹(hit groups)” 수를 지정. Default: 2 |
결과
-

kraken.log 파일 내용. Kraken2 실행에 대한 간단한 통계치. Kraken DB에 등록된 종으로 분류된(classified) read 수와 종을 찾지 못한(unclassfied) read 수를 나타냄.
-
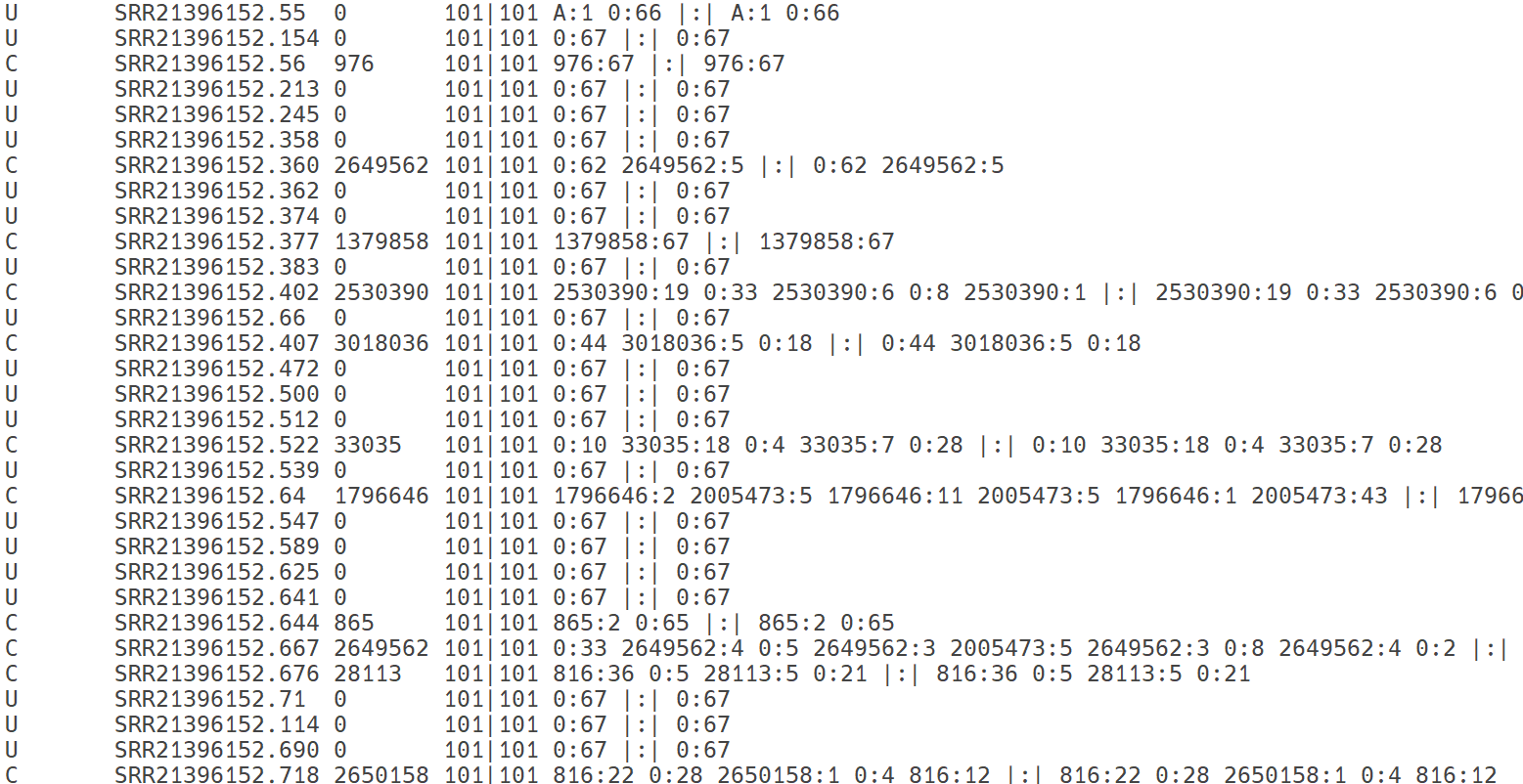
kraken.txt 파일 내용. 전체 Kraken2 분류 결과를 보여 줌. 첫번째 열부터 차례로 분류 여부(C: Classified, U: Unclassified), 서열명, 분류군 ID(NCBI 기준), 매핑 정보를 나타냄.
-
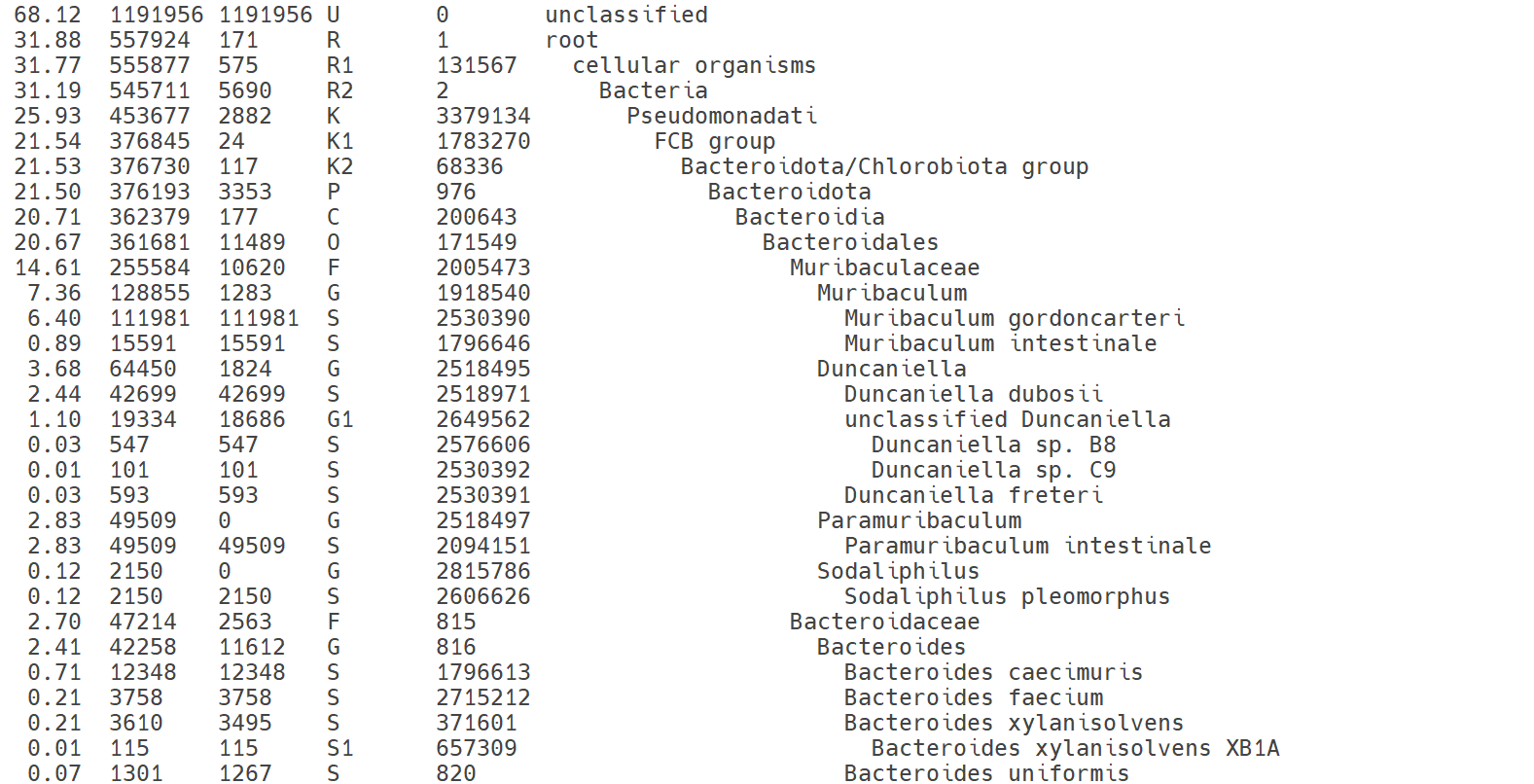
report.txt 파일 내용. 각 분류군 별로 얼마나 많은 read가 분포하는지 요약함. 첫 번째 열부터 차례로 read 수의 백분율, 해당 분류군 또는 하위 분류군에 할당된 read 수, 해당 분류군에만 할당된 read 수, 분류군 ID, 분류군 이름을 나타냄.
krona
Krona는 Kraken2와 같은 분류 도구로부터 생성된 결과를 기반으로, 각 분류군의 상대적인 비율과 계통 구조를 직관적으로 시각화하는 도구입니다. Krona는 계통 분류 결과를 다층 원형(hierarchical circular) 형태의 인터랙티브 HTML 시각화로 표현하여, 각 생물군의 비율, 서열 수, 상위·하위 관계를 한 눈에 탐색할 수 있도록 지원합니다.
생성된 HTML 결과 파일은 별도의 프로그램 설치 없이 웹 브라우저에서 직접 열어볼 수 있으며, 샘플 내 주요 미생물 군집 구성이나 잠재적 오염 여부를 시각적으로 확인하는 품질 관리(QC) 용도로 사용됩니다.
실행 명령어 예시
Krona.sh input_dir=./kraken2/output output_dir=./krona/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./kraken2/output | 분석에 사용될 fastq 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./krona/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
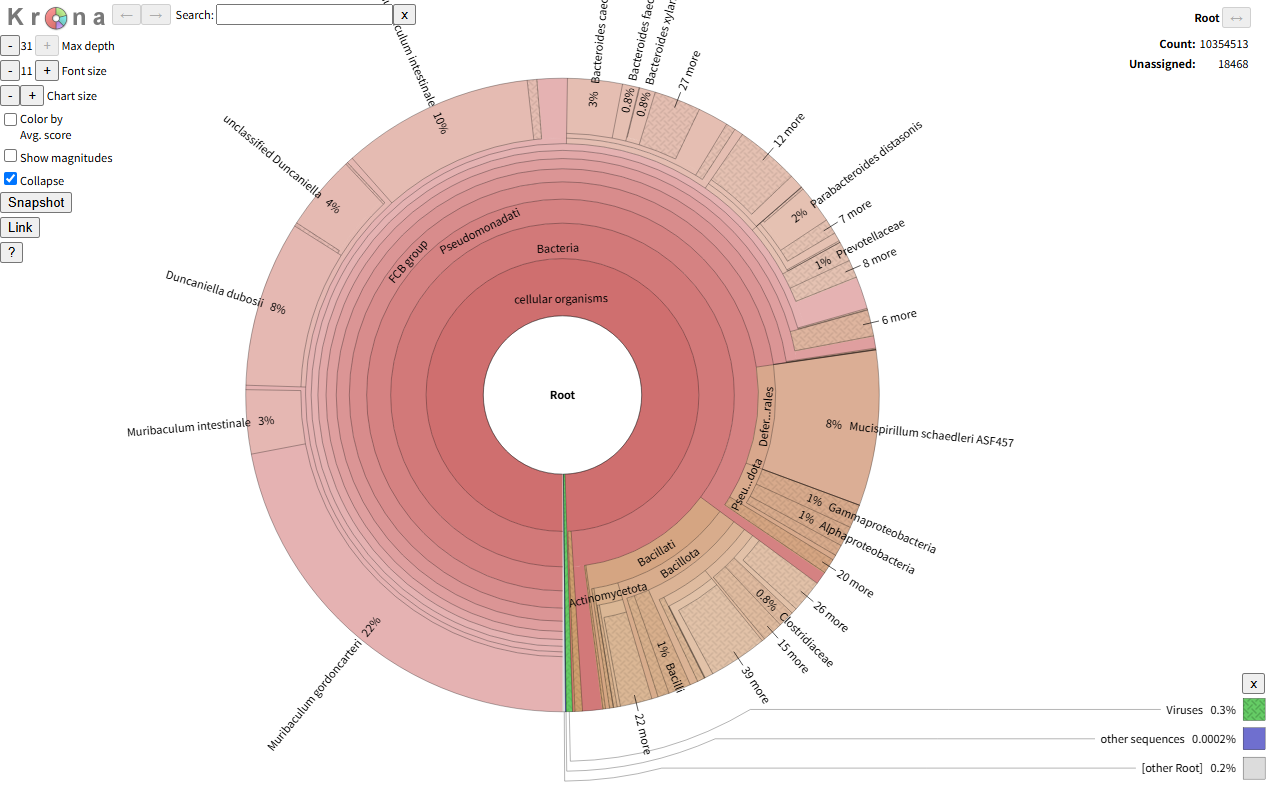
krona plot이 담긴 결과 html 파일. 각 분류 레벨 별 미생물 종 분포를 확인할 수 있음. 클릭 시 반응하는 인터랙티브 단일 파일 웹페이지로, 웹 브라우저에서 실행됨.
bbnorm
균질화(Normalization) 단계는 메타게놈 시퀀싱 데이터의 리드(read) 커버리지(coverage)를 균질화하여 어셈블리의 효율을 높이고 계산 자원을 절약하기 위한 전처리 단계입니다. 일반적으로 시퀀싱 데이터에는 특정 유전체 영역이 과도하게 많이 읽히는(high coverage) 경우가 발생하는데, 이러한 불균형은 어셈블리 과정에서 메모리 사용량 증가와 조립 품질 저하를 초래할 수 있습니다. BBNorm은 각 서열의 빈도(K-mer 빈도 기준)을 계산하여, 높은 커버리지 영역의 리드 수를 감소시키고 낮은 커버리지 영역의 서열은 유지함으로써, 데이터의 복잡도를 줄이면서 정보 손실을 최소화합니다.
이 과정은 특히 MEGAHIT 같은 대규모 메타게놈 어셈블러를 사용하기 전에 수행되며, 불필요하게 중복된 리드를 제거하여 조립 속도와 정확도를 향상시킵니다. 또한 normalization을 통해 과도하게 표현된 개체군에 의한 편향을 줄여, 상대적으로 희귀한 미생물 서열이 어셈블리에 더 균등하게 반영될도록 돕습니다.
주요사항
- BBNorm은 Java 기반 NGS 데이터 처리 도구 모음인 BBMap(BBTools) 툴킷에 포함된 구성 요소 중 하나임.
실행 명령어 예시
bbnorm.sh input_dir=./bowtie2_unmapped/output output_dir=./bbnorm/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./bowtie2_unmapped/output | 분석에 사용될 전처리된 fastq 파일들이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./bbnorm/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 | |
| Option | Integer | target | 100 | 리드의 평균 커버리지를 지정된 목표 깊이로 균질화하는 값 (Default: 100) | |
| Option | Integer | mindepth | 5 | 지정된 최소 커버리지 이하의 리드는 제거하지 않고 유지하는 기준값 (Default: 5) |
결과
-
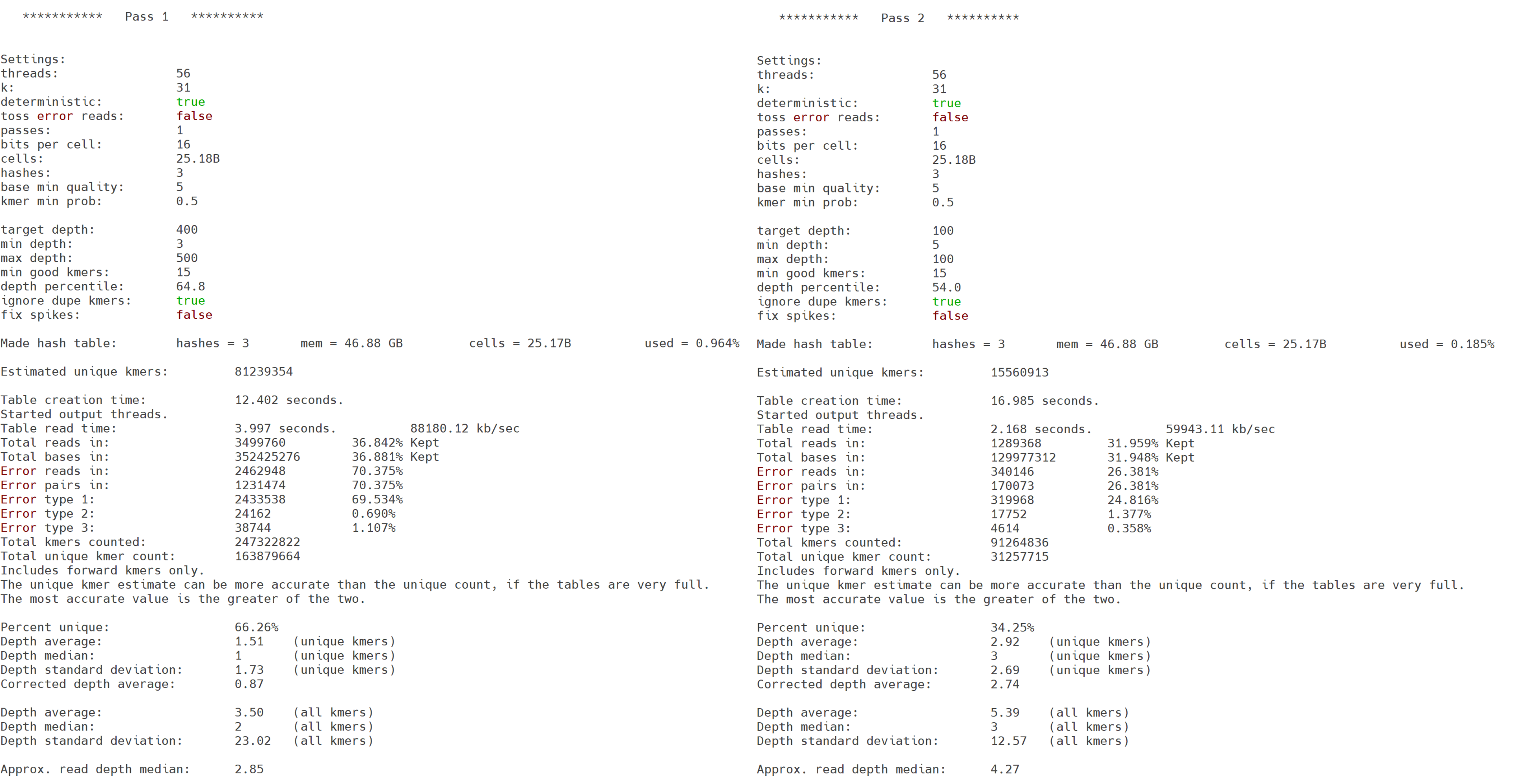
BBNorm 실행 로그를 통해 K-mer 분포, 리드 유지 비율, 고유 서열 복잡도, 평균 커버리지 등 정규화 과정의 세부 통계와 처리 과정을 확인할 수 있음.
megahit
Megahit 단계는 메타게놈 시퀀싱 데이터로부터 고품질의 contig(연속 서열)를 조립(assembly) 하는 핵심 과정입니다. 메타게놈 데이터는 다양한 생물종의 서열이 혼합되어 있어 복잡도가 매우 높으며, 이를 효율적으로 조립하기 위해서는 대용량 데이터를 빠르고 안정적으로 처리할 수 있는 알고리즘이 필요합니다. Megahit은 이러한 대규모 메타게놈 데이터를 위한 초고속 de Bruijn graph 기반 어셈블러로, 다중 K-mer 점진 확장 기법(iterative multi-k-mer strategy)을 사용하여 짧은 리드를 긴 contig으로 재구성합니다.
출력 결과로 생성되는 final.contigs.fa 파일은 이후 binning(예: MetaBAT2, MaxBin2, CONCOCT) 단계 및 refinement (예: DAS_Tool)의 입력으로 활용됩니다. 이 단계는 메타게놈 분석 파이프라인 전체의 품질과 효율을 결정하는 핵심 절차로, 전처리된 고품질 리드 입력이 MEGAHIT 조립 결과에 직접적인 영향을 미칩니다.
실행 명령어 예시
megahit.sh \ input_dir=./bbnorm/output \ output_dir=./megahit/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./bbnorm/output | 분석에 사용될 fastq 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./megahit/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-

final.contigs.fa 파일 내용. 기존 read를 MEGAHIT으로 assembly한 최종 결과물
quast_megahit
metaQUAST(Metagenome Assembly Quality Assessment Tool)는 복합 미생물 군집의 메타게놈 어셈블리 품질을 평가하기 위해 개발된 QUAST의 확장형 버전으로, MEGAHIT이나 metaSPAdes 등의 결과를 대상으로 합니다. 다양한 종이 혼합된 시료의 특성을 고려해 reference genome 없이도 contig 기반 통계(N50, GC%, contig 길이 분포 등)를 계산하며, 내부적으로 RefSeq 데이터베이스에서 유사 reference를 자동 탐색하는 기능이 활성화되어, contig 일부를 BLAST 검색합니다. 이 과정에서 alignment를 수행해 각 contig가 어떤 종인지 추정하고, Krona 그래프 형태의 대략적인 taxonomic 구성비를 산출합니다. 이를 통해 metaQUAST는 어셈블리의 전반적인 품질뿐만 아니라 잠재적 오염과 종 구성 정보를 함께 제공합니다.
주요사항
- metaQUAST는 QUAST에서 확장되어 개발된 메타게놈 전용 품질 평가 도구임
실행 명령어 예시
quast_megahit.sh \ input_dir=./megahit/output \ output_dir=./quast_megahit/output
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./megahit/output | 분석에 사용될 assembled fasta 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./quast_megahit/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
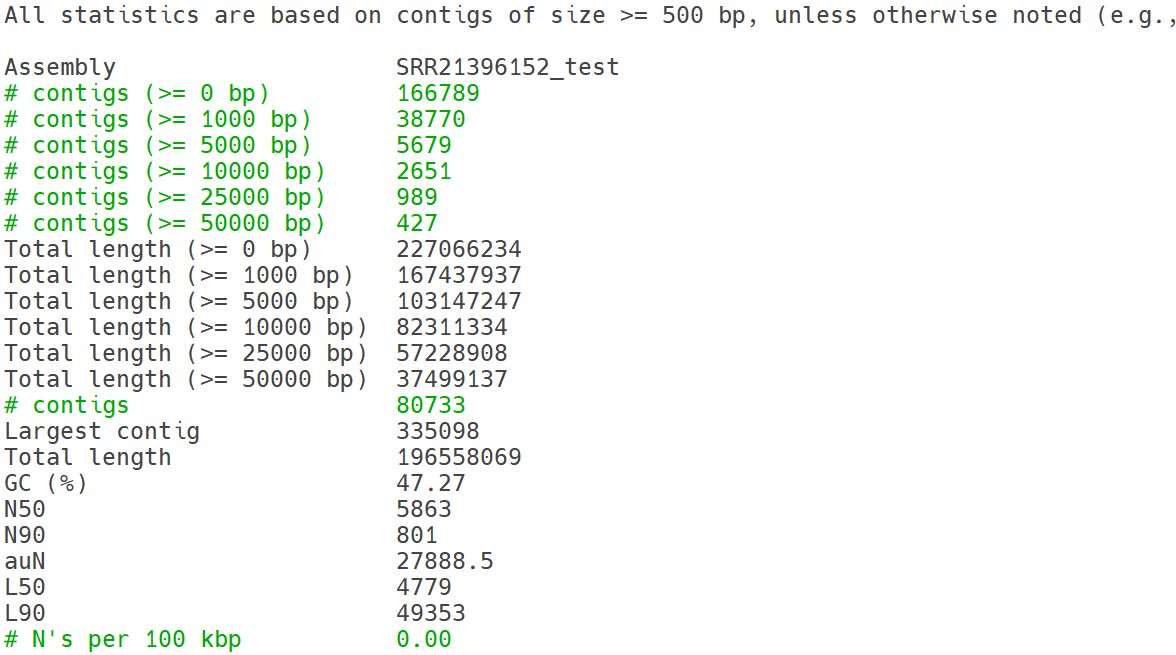
report.txt 파일 내용. 입력 데이터의 contig 수, GC 함량 등 기본적인 수치와 assmebly의 완성도를 평가할 N50, L50 등의 품질 수치를 담고 있음.
-
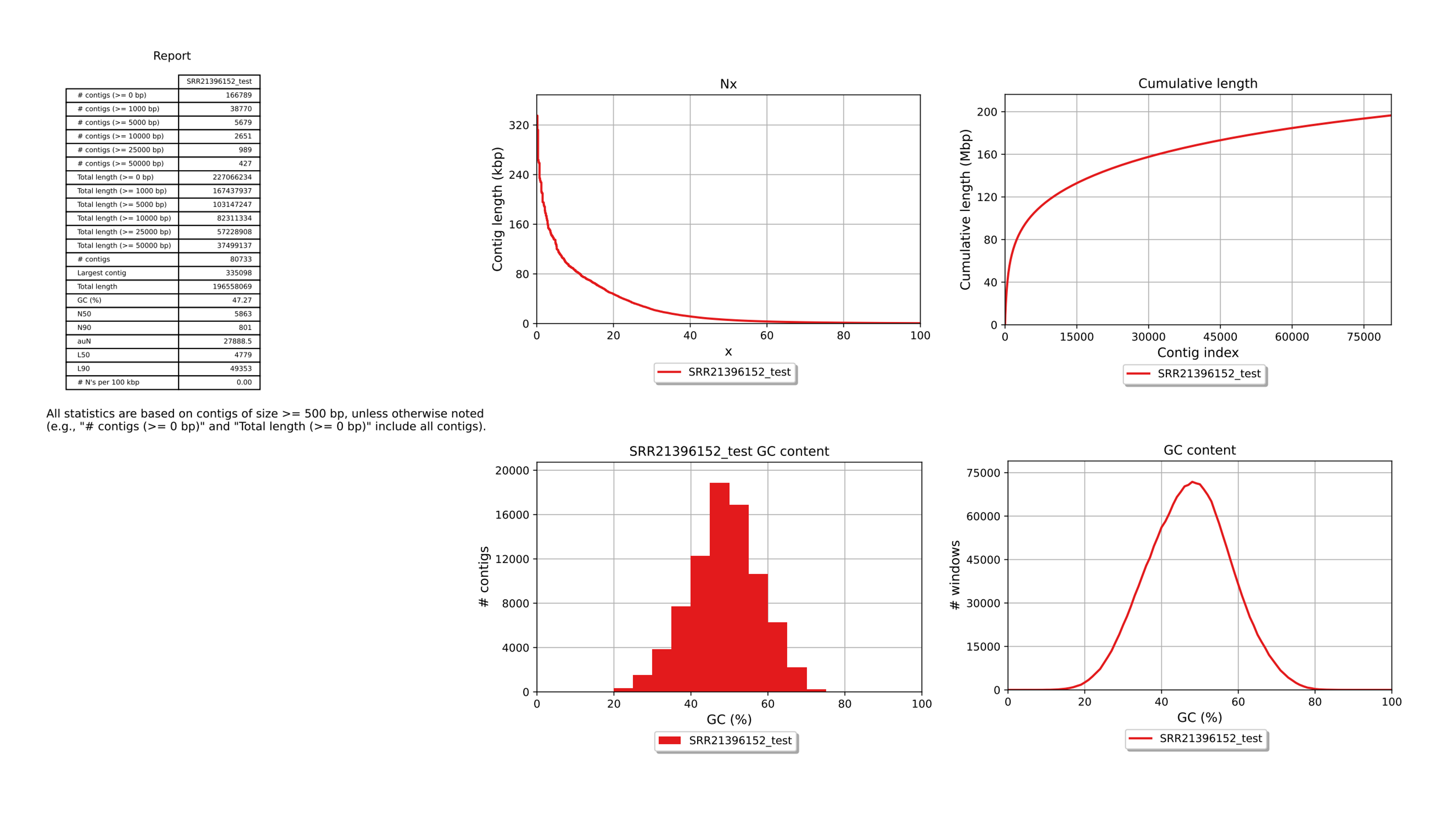
report.pdf 파일 내용. report.txt의 수치 및 이를 시각화한 그래프를 담고 있음.
-
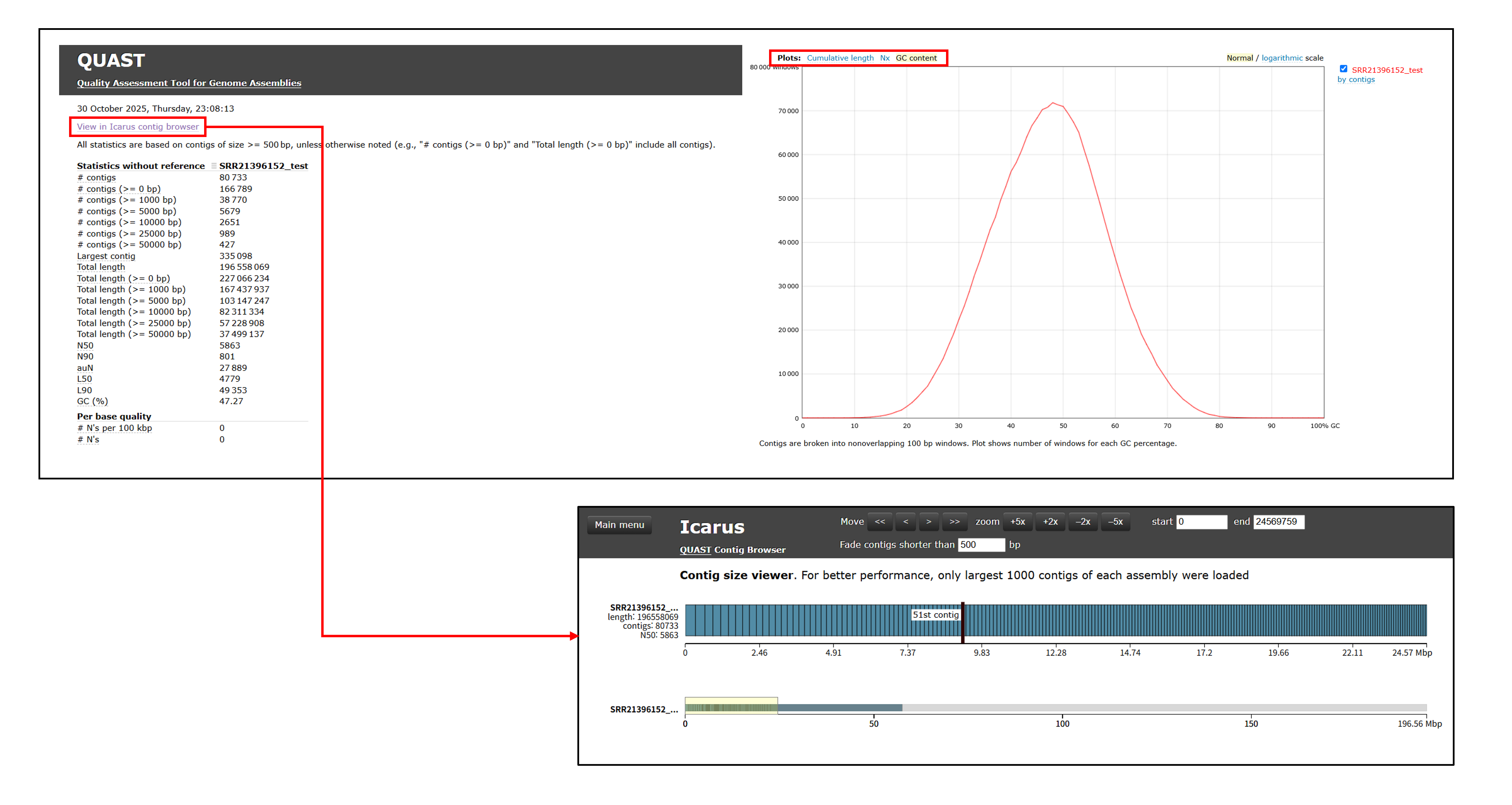
report.html 파일 내용. report.txt의 통계 수치 및 이를 시각화한 그래프, 그리고 contig 구조에 대한 genome map을 제공하고 있음.
prodigal
Prodigal (Prokaryotic Dynamic Programming Gene-finding Algorithm)은 어셈블러를 통해 생성된 유전체 조립 결과(contigs 또는 scaffolds)를 입력으로 하여, 원핵생물의 유전체 내 단백질 암호화 유전자(coding DNA sequence, CDS)를 예측하는 도구입니다. Prodigal은 동적 프로그래밍 기반 알고리즘을 사용하여 염기서열 내의 ORF(Open Reading Frame)를 탐색하고 각 유전자의 위치, 방향, 시작·종결 코돈, 단백질 서열 등을 예측합니다.
Prodigal 실행 후에는 여러 형식의 결과 파일이 생성됩니다.
- 각 contig 내에서 예측된 유전자의 위치 정보(좌표)와 기본 구조(annotation feature)를 포함하는 파일 (.gff)
- 예측된 CDS의 염기서열을 FASTA 형식으로 저장한 파일 (.ffn 또는 .fna),
- 예측된 CDS를 번역(translation)하여 단백질 서열(protein sequence)로 저장한 파일(.faa)
주요사항
- Prodigal은 Prokka, CheckM2, GTDB-Tk 등 기능 분석 도구에서 내부적으로 자동 호출되어 유전자 예측에 활용됨
- 메타게놈 분석에서 어셈블리 이후 prodigal 단계는 품질 관리(QC)와 주석 (annotation)을 준비하기 위한 사전 유전자 예측 단계로 활용됨 (mode: meta)
실행 명령어 예시
prodigal.sh input_dir=./megahit/output output_dir=./prodigal/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./megahit/output | 분석에 사용될 assembled fasta 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./prodigal/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 | |
| Option | String | output_format | gff | 결과 파일의 출력 형식 지정. gbk, gff(default), sco 중 택일 | |
| Option | String | mode | meta | 입력 데이터의 특성에 따라 분석 모드를 지정. single, meta(default) 중 택일 | |
| Option | Integer | translate | 11 | 코돈을 아미노산으로 번역 시 사용할 유전 부호 표 번호 지정. Default: 11 (Bacterial, Archaeal and Plant Plastid Code) |
결과
-
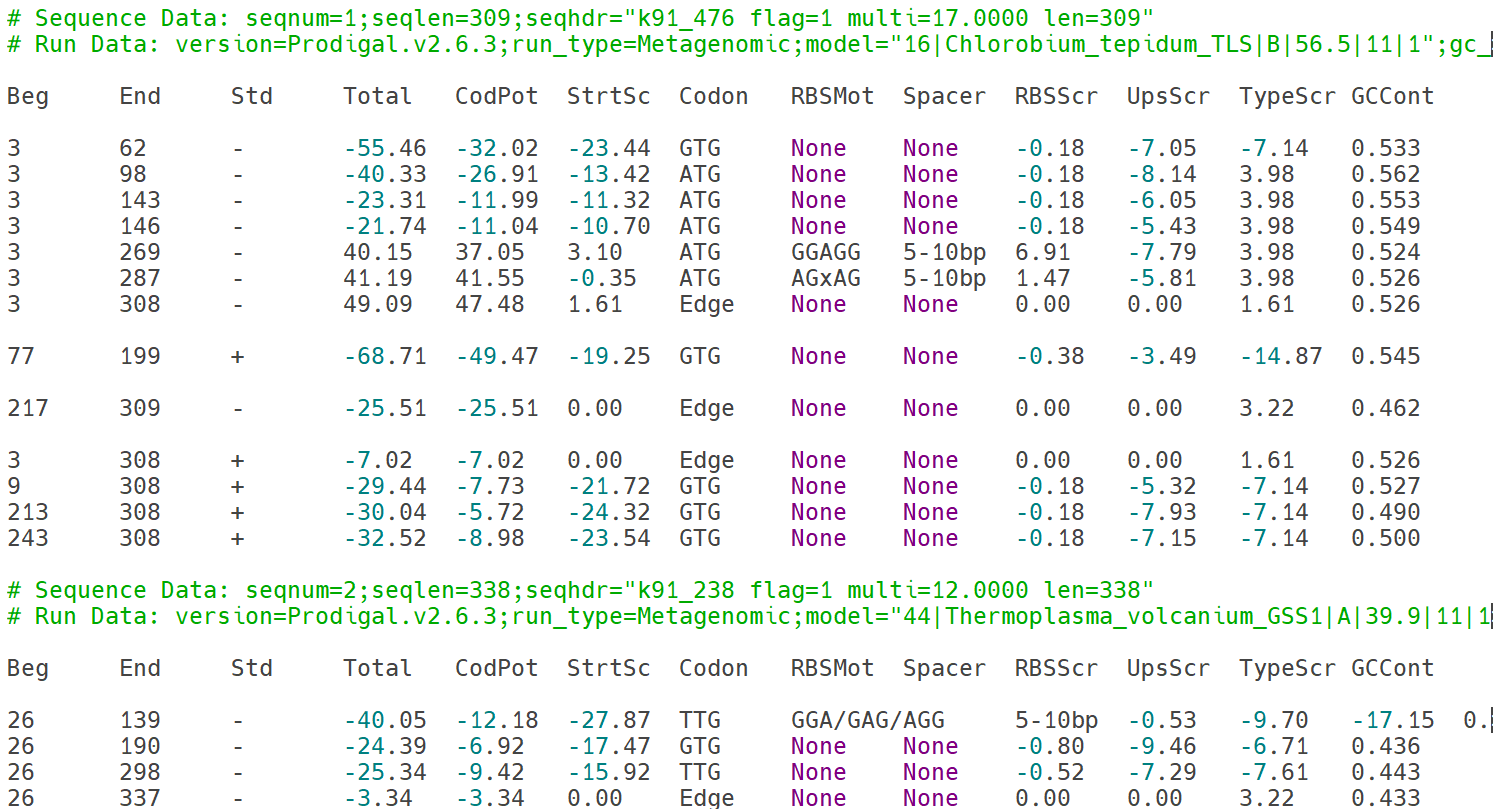
_all.txt 파일 내용. 예측된 유전자 및 전체 ORF (Open Reading Frame)에 대해 각 수치를 나타낸 표. 첫번째 열부터 차례로 ORF의 시작 코돈, 종료 코돈, DNA 가닥 방향, 유전자 후보 점수(점수가 높을수록 실제 유전자일 가능성이 높음) 등을 나타냄.
-
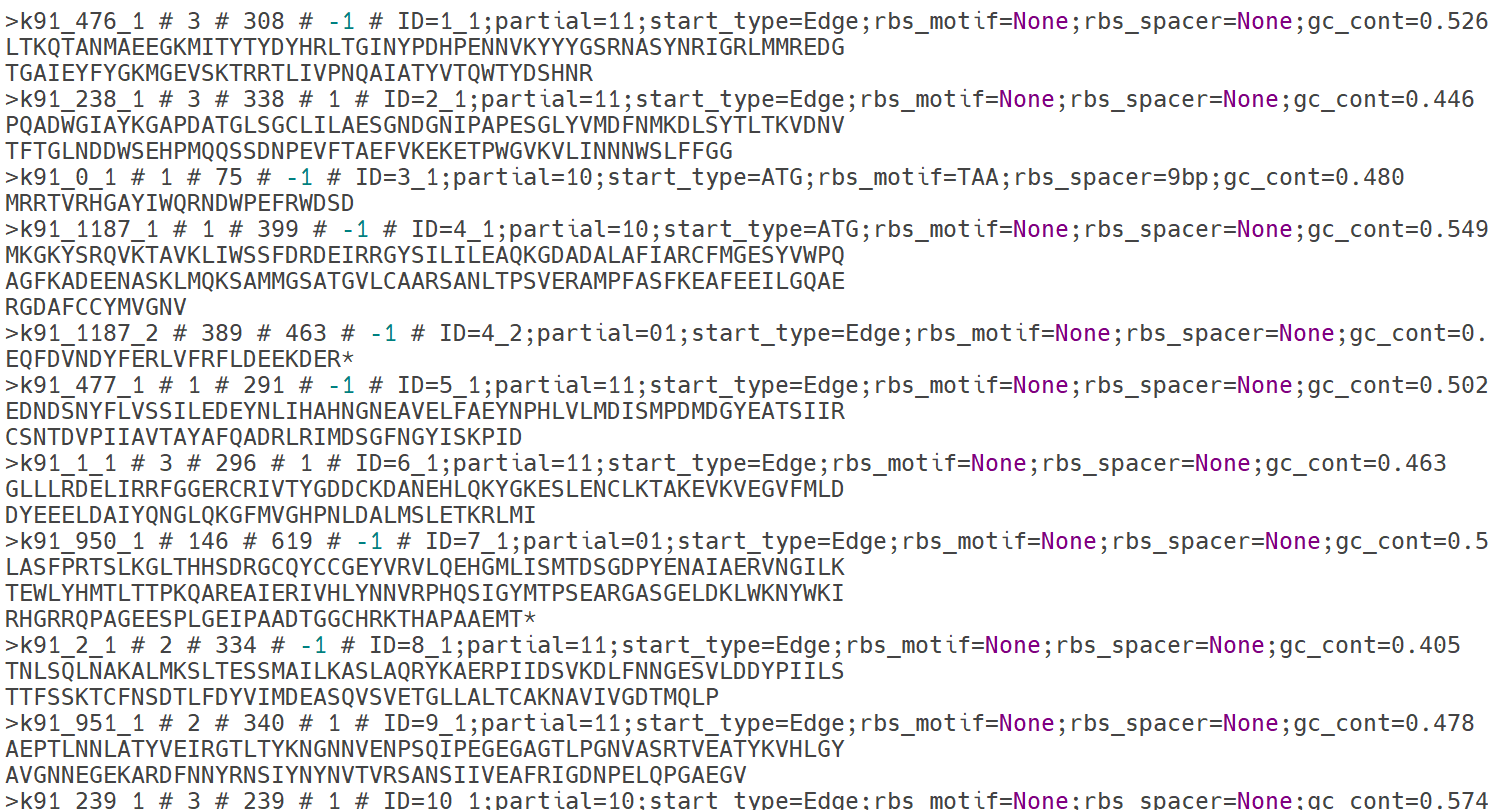
.faa 파일 내용. 예측된 각 유전자 서열을 단백질 아미노산 서열로 번역한 정보를 담은 FASTA 형식의 파일.
-
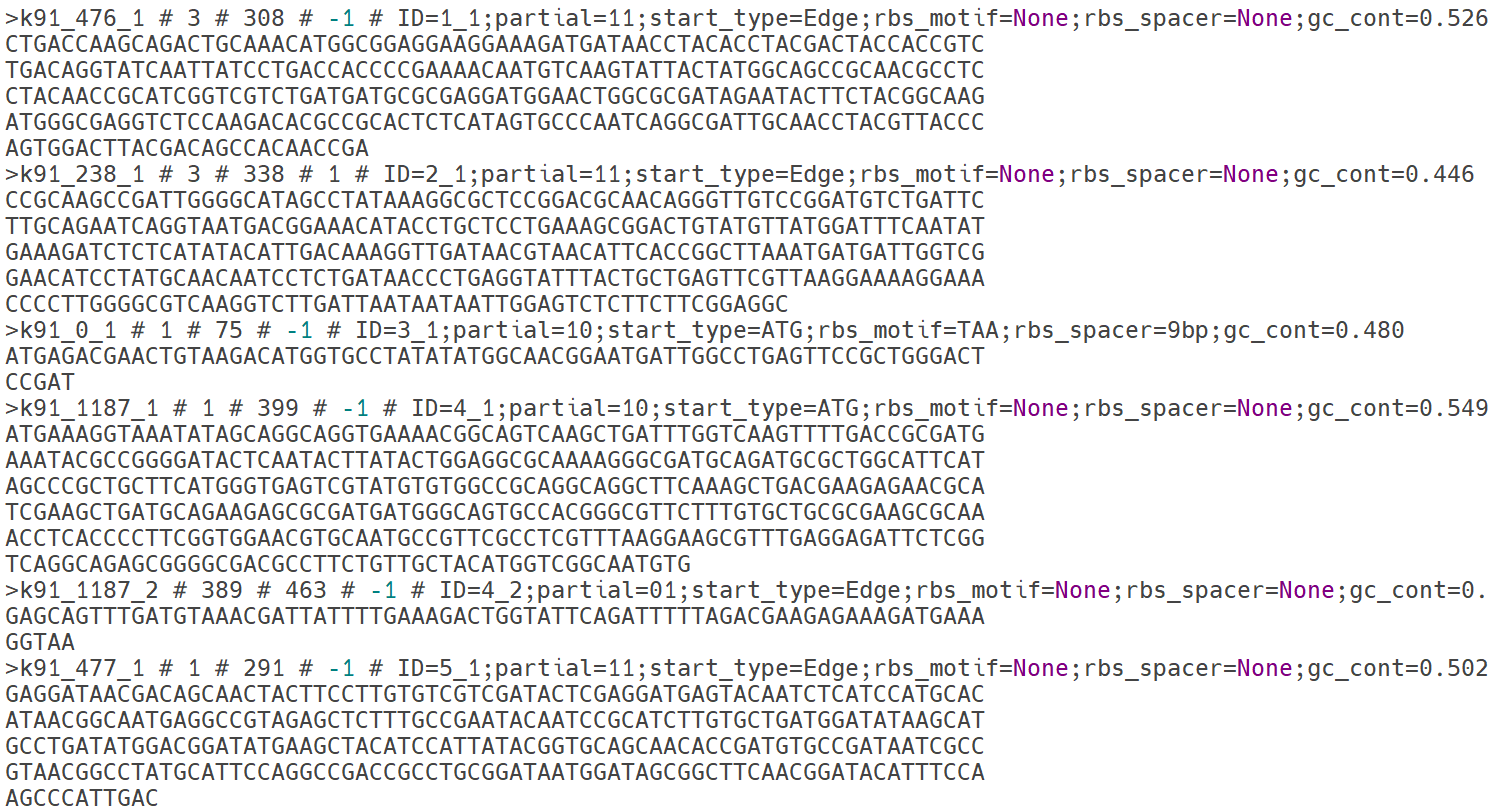
.fna 파일 내용. 예측된 각 유전자 서열 (Coding Sequence, CDS) 정보를 담은 FASTA 형식의 파일.
-

.gff 파일 내용. gff는 대표적인 서열 주석 파일 형식으로, 예측된 각 유전자에 대해 서열 이름, 시작점, 끝점, 방향과 같은 위치 정보와 score, start codon type 등의 상세 정보가 포함됨.
bowtie2_reAlign
Bowtie2는 Johns Hopkins University의 Ben Langmead와 Steven Salzberg가 개발한 플랫폼으로, 전 세계 주요 게놈 센터와 바이오인포매틱스 분야에서 서열 정렬 분야의 표준 참조로 인정받고 있는 핵심 도구입니다. 이 도구는 기존 정렬 도구들의 한계를 극복하기 위해 FM-index(Full-text Minute-space index) 기술과 BWT(Burrows-Wheeler Transform) 알고리즘을 결합한 획기적인 색인 구조를 구현하여, 인간 전체 게놈과 같은 거대한 참조 서열에서도 메모리 효율성과 검색 속도를 동시에 최적화한 혁신적 솔루션을 제공합니다. Bowtie2의 가장 큰 기술적 혁신은 갭을 허용하는 정렬(gapped alignment)과 지역 정렬(local alignment) 기능을 완벽하게 지원한다는 점입니다. 이는 ChIP-seq 분석에서 핵심적으로 중요한데, 크로마틴 면역침전 과정에서 발생할 수 있는 DNA 단편화나 시퀀싱 오류를 지능적으로 처리하면서도 multi-mapping 리드의 정확한 분류를 통해 반복 서열 영역에서의 false positive 신호를 효과적으로 차단합니다. 특히 MAPQ(Mapping Quality) 점수 계산 시스템을 통해 각 정렬의 신뢰도를 정량적으로 평가하여, 후속 MACS2 피크 호출에서 고유하게 매핑된 고신뢰도 리드만을 선별적으로 활용할 수 있게 합니다.
주요사항
- $input_dir에는 Single-end와 paired-end FASTQ 파일 모두 처리 가능합니다. 이는 fastq 또는 fq 확장자 앞에 있는 “_R1/2”, “.R1/2”, “_1/2” 형식의 존재 여부로 single 및 paired end 여부를 자동으로 처리합니다.
- 기존 Bowtie2의 툴을 기반으로 기능이 일부 고도화되었습니다. reference file 입력으로 Megahit의 결과 파일(final.contigs.fa)을 자동 인식하도록 개선되었습니다.
실행 명령어 예시
bowtie2_reAlign.sh \ input_dir=./bowtie2_unmapped/output \ reference_genome_dir=./bowtie2_realign/output \ output_dir=./bowtie2_realign/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./bowtie2_unmapped/output | 분석에 사용될 전처리된 fastq 파일들이 있는 디렉터리 경로 | |
| Input | Folder | reference_genome_dir | ./megahit/output | 분석에 사용될 참조 서열 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./bowtie2_realign/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
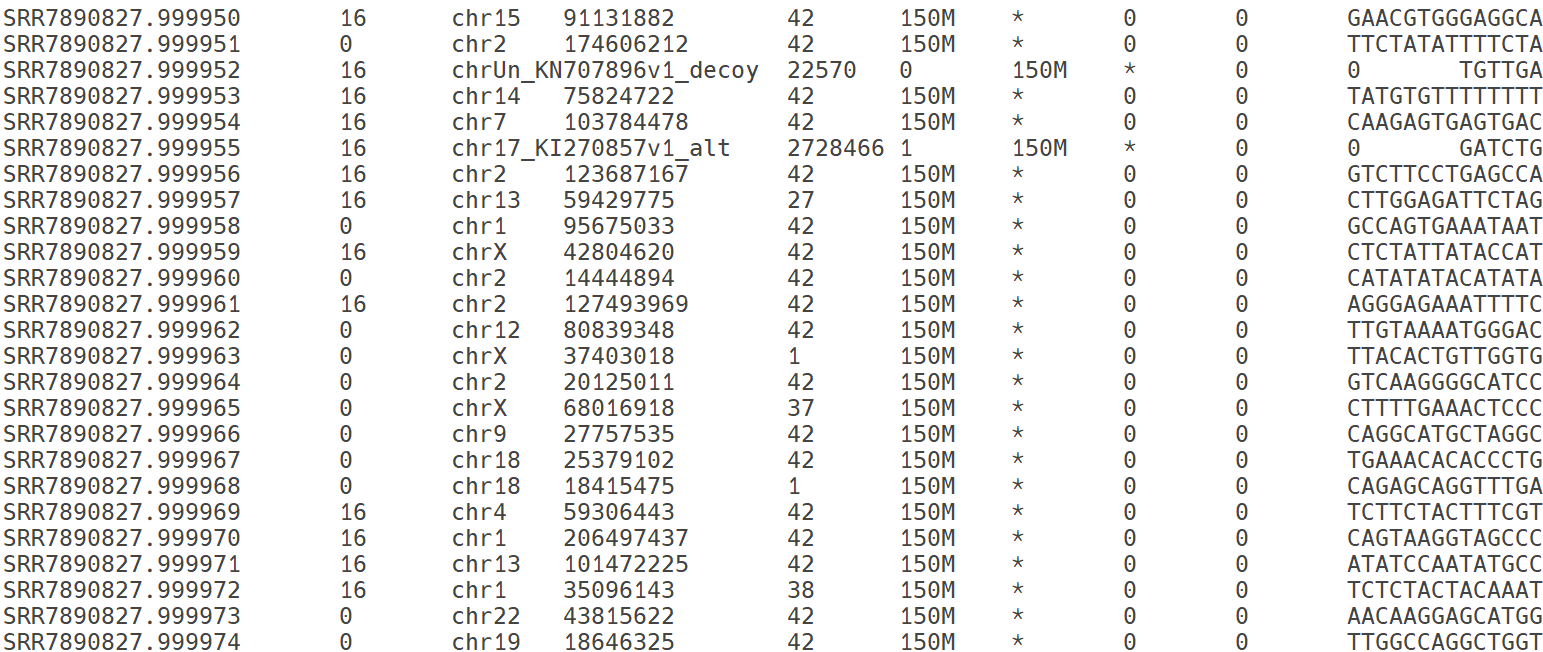
SAM 파일의 본문 영역. mapping 정보를 제공. 제시된 이미지의 각 열은 다음과 같은 정보를 제공함: read 이름, 정렬 상태, 참조서열 이름, 정렬의 왼쪽 위치, 정렬 품질, 정렬 상태, paried-end의 다음 read 정렬 정보, paried-end의 두 read 사이의 거리, read의 염기서열 정보를 제공하고 있음.
-
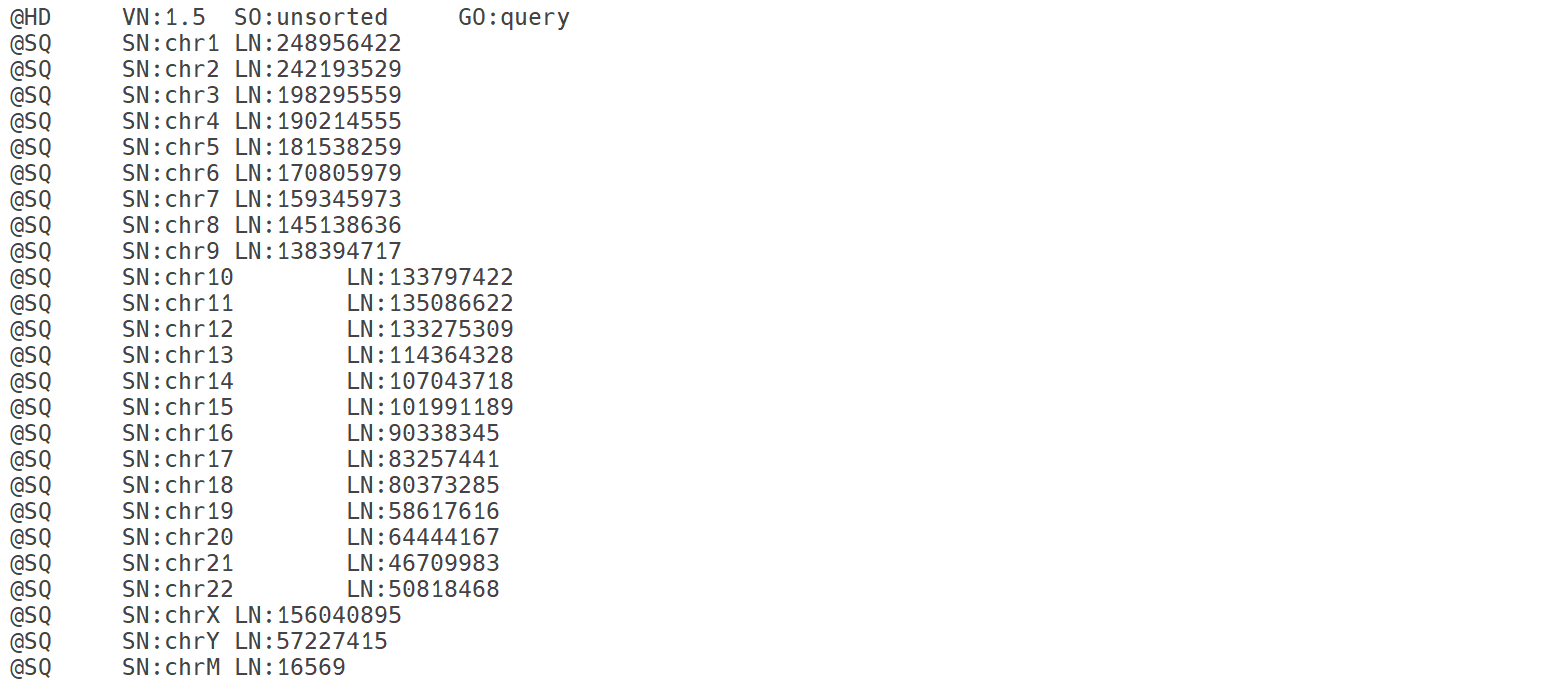
SAM 파일의 header 영역. @HD: header 영역 전체를 정의. VN: SAM 파일의 포맷 버전 SO: 정렬 방식 GO: 그룹화 방식. query는 read 이름 별로 그룹화되었음을 의미. @SQ: 참조 서열에 대한 정보를 제공함. 이 경우 사람 genome의 각 염색체에 대한 정보를 제공함. @RG: read 그룹의 정보를 제공함. @PG: 프로그램 그룹의 정보로, 정렬 도구 등의 정보를 제공함.
SAMtools_sort
SAMtools는 고처리량 시퀀싱 데이터를 조작하고 분석하기 위해 설계된 다목적 도구 모음으로, 특히 SAM, BAM, CRAM 형식의 정렬 데이터를 처리하는 데 사용됩니다. 이 도구는 변이 호출, 정렬 데이터 확인, 품질 관리 등 다양한 작업에 널리 활용됩니다. SAMtools는 인덱싱, 정렬, 병합, 필터링 등 정렬 파일 처리에 필요한 다양한 기능을 지원하며, 유전체 분석 파이프라인에서 필수적인 도구입니다.SAMtools sort 명령은 SAM, BAM, CRAM 파일을 지정된 기준에 따라 정렬합니다. 기본적으로 좌표(coordinate) 기준으로 정렬하며, -n 옵션을 사용하면 쿼리 이름(query name) 기준으로 정렬합니다. 정렬 순서는 SAM 파일 헤더의 @HD 태그 내 SO 필드에 표시됩니다. 좌표 정렬은 @HD SO:coordinate, 쿼리 이름 정렬은 @HD SO:queryname으로 헤더에 기록됩니다.
좌표 정렬 (Coordinate Sorting)의 경우, 읽기는 다음과 같은 순서로 정렬됩니다:
1. 참조 시퀀스 이름(RNAME): @SQ 태그에 정의된 참조 시퀀스 사전의 순서를 따릅니다.
2. 가장 왼쪽 매핑 위치(POS): 동일한 RNAME 내에서 읽기의 시작 위치를 기준으로 정렬됩니다.
3. REVERSE 플래그: POS가 동일한 경우, 순방향 가닥(forward strand, REVERSE 플래그 0)이 역방향 가닥(reverse strand, REVERSE 플래그 1)보다 먼저 옵니다.
이후 추가적인 동점이 있는 경우, 정렬 순서는 입력 데이터의 순서를 유지할 수 있습니다.
쿼리 이름 정렬 (Query Name Sorting)의 경우, -n 옵션을 사용하며 읽기는 다음과 같은 순서로 정렬됩니다:
1. 쿼리 이름(QNAME): 자연스러운 순서(natural order)로 정렬되며, 문자열 내 숫자 부분은 수치적으로 비교됩니다 (예: "read9"는 "read10"보다 먼저).
2. READ1/READ2 플래그: 동일한 QNAME을 가진 읽기는 첫 번째 읽기(READ1, 플래그 0x40)가 두 번째 읽기(READ2, 플래그 0x80)보다 먼저 옵니다.
3. 정렬 유형: READ1/READ2가 동일한 경우, 주 정렬(primary alignment)이 먼저 오고, 그 다음 보조 정렬(supplementary alignment), 2차 정렬(secondary alignment) 순으로 정렬됩니다.
남은 동점은 입력 데이터의 원래 순서를 따릅니다. 따라서 SAMtools는 유전체 데이터를 효율적으로 처리하고 분석하는 데 중요한 역할을 하며, 특히 정렬 작업에서 sort 도구가 유용하게 사용됩니다. Bio-Express의 Whole Genome Sequencing Pipeline 분석과정에서 SAMtools sort는 SAM파일을 입력 데이터로 사용하여 BAM 파일을 출력합니다.
실행 명령어 예시
$PROGRAM_DIR/samtools sort -@ 6 –O bam —write-index –o $OUTPUT_BAM $INPUT_BAM
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | /path/to/input_dir | Sorting이 필요한 BAM 파일을 찾을 디렉토리 | |
| Output | Folder | output_dir | /path/to/output_dir | Sorting이 완료된 BAM 파일을 저장할 디렉토리 |
결과
-

좌표 기준 정렬 전 상태. 모든 레코드의 RNAME(3열)은 chr1이며, POS(4열) 순서가 10000 → 10002 → 10001 → 10003 처럼 뒤섞여 있음. 둘째~셋째 줄의 chr2 표기는 RNEXT(7열)로, 짝 리드의 위치 정보일 뿐 현재 레코드의 정렬 참조는 아님.
-

좌표 기준 정렬된 상태. 모든 레코드의 RNAME(3열)이 chr1이며, POS(4열)이 10000 → 10001 → 10002 → 10003 순으로 오름차순. 둘째 줄의 chr2는 RNEXT(7열)로, paired 리드가 chr2에 매핑됨을 의미(해당 레코드는 여전히 chr1에 정렬). RNEXT가 =인 경우는 paired 리드가 같은 염색체(여기서는 chr1)에 정렬되어 있다는 뜻.
concoct
CONCOCT (Clustering cONtigs with COverage and ComposiTion)는 메타게놈 시퀀싱 데이터를 기반으로 조립된 contig들을 염기서열 조성(nucleotide composition)과 서열의 샘플별 coverage 패턴의 유사성을 이용해 군집화(binning)하는 도구입니다. Gaussian Mixture Model(GMM) 통계 모델을 적용하여, contig들을 잠재적인 개별 미생물 유전체 단위(bin)로 분류하며, 이를 통해 서로 다른 미생물종이 혼합된 시료로부터 배양되지 않은 미생물의 게놈을 복원할 수 있습니다. Coverage 계산에는 Bowtie2 및 SAMtools의 결과 파일이 입력으로 사용됩니다.
주요사항
- CONCOCT는 여러 하위 스크립트(cut_up_fasta, coverage_table, clustering, extract_bins 등)로 구성된 모듈형 파이프라인을 통해 contig 분할, 커버리지 계산, 클러스터링 및 bin 추출 단계를 순차적으로 수행함
- 본 프로그램은 후속 통합 분석에 사용할 수 있도록 DAS Tool의 입력 요구사항에 맞추어 결과 파일을 변환함
실행 명령어 예시
Concoct.sh input_dir=./SAMtools_sort/output fasta_dir=./megahit/output output_dir=./Concoct/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./SAMtools_sort/output | 분석에 사용될 정렬된 BAM 파일이 있는 디렉터리 경로 | |
| Input | Folder | fasta_dir | ./Megahit/output | 분석에 사용될 assembled fasta 파일이 있는 디렉터리 경로 | |
| Output | String | output_dir | ./Concoct/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
.png)
concoct_contigs2bin.tsv 파일 내용. CONCOCT 분석 결과 각 conitg와 그에 부여된 최종 genome bin ID를 매핑한 파일.
-
.png)
fasta_bins_merged/ 디렉토리의 파일과 해당 파일의 구조. 이 디렉토리는 CONCOCT 분석 결과 생성된 genome bin에 대한 FASTA 파일들이 위치해 있음. 각 파일에는 해당 bin에 소속된 contig 서열 정보가 담겨 있음.
-
.png)
_cutted_contigs.bed 파일 내용. 길이 기준을 충족해 분석에 사용된 contig 서열의 위치 정보를 포함하는 BED 형식 파일.
-
.png)
_corverage_table.tsv 파일 내용. 각 contig의 시퀀싱 커버리지 수치에 대한 표로, 동일한 샘플에서 유래한 contig는 유사한 커버리지 수치를 가진다는 사실을 기반으로 binning 과정에서 핵심 수치로 사용됨.
metabat2
MetaBAT2(Metagenome Binning with Abundance and Tetra-nucleotide frequencies 2)는 메타게놈 어셈블리 결과로부터 생성된 contig들을 염기서열 조성(tetranucleotide frequency, TNF)과 샘플별 coverage 정보를 바탕으로 자동 분류(binning)하는 도구입니다. Bayesian 확률 모델을 이용해 contig 간의 유사도를 계산하고, 동일한 유래(genome)로 추정되는 contig들을 하나의 bin으로 군집화합니다. 이를 통해 복합 미생물 군집으로부터 개별 미생물 유전체(MAG)를 재구성할 수 있습니다. MetaBAT2는 속도와 정확도가 높아 대규모 샘플의 병렬 처리에 적합하며, coverage 정보는 Bowtie2 및 SAMtools 결과 파일을 기반으로 계산됩니다.
주요사항
- MetaBAT2는 jgi_summarize_bam_contig_depths를 이용해 샘플별 BAM 파일로부터 contig 단위의 커버리지를 계산한 후, metabat2를 통해 염기서열 조성과 커버리지 정보를 기반으로 binning을 수행하는 2단계 파이프라인 구조로 동작함
- 본 프로그램은 후속 통합 분석에 사용할 수 있도록 DAS Tool의 입력 요구사항에 맞추어 결과 파일을 변환함
실행 명령어 예시
metabat2.sh \ input_dir=./SAMtools_sort/output \ fasta_dir=./megahit/output \ output_dir=./Metabat2/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./SAMtools_sort/output | 분석에 사용될 정렬된 BAM 파일이 있는 디렉터리 경로 | |
| Input | Folder | fasta_dir | ./megahit/output | 분석에 사용될 assembled fasta 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./Metabat2/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
.png)
metabat2_contigs2bin.tsv 파일 내용. Metabat2 분석 결과 각 conitg와 그에 부여된 최종 genome bin ID를 매핑한 파일.
-
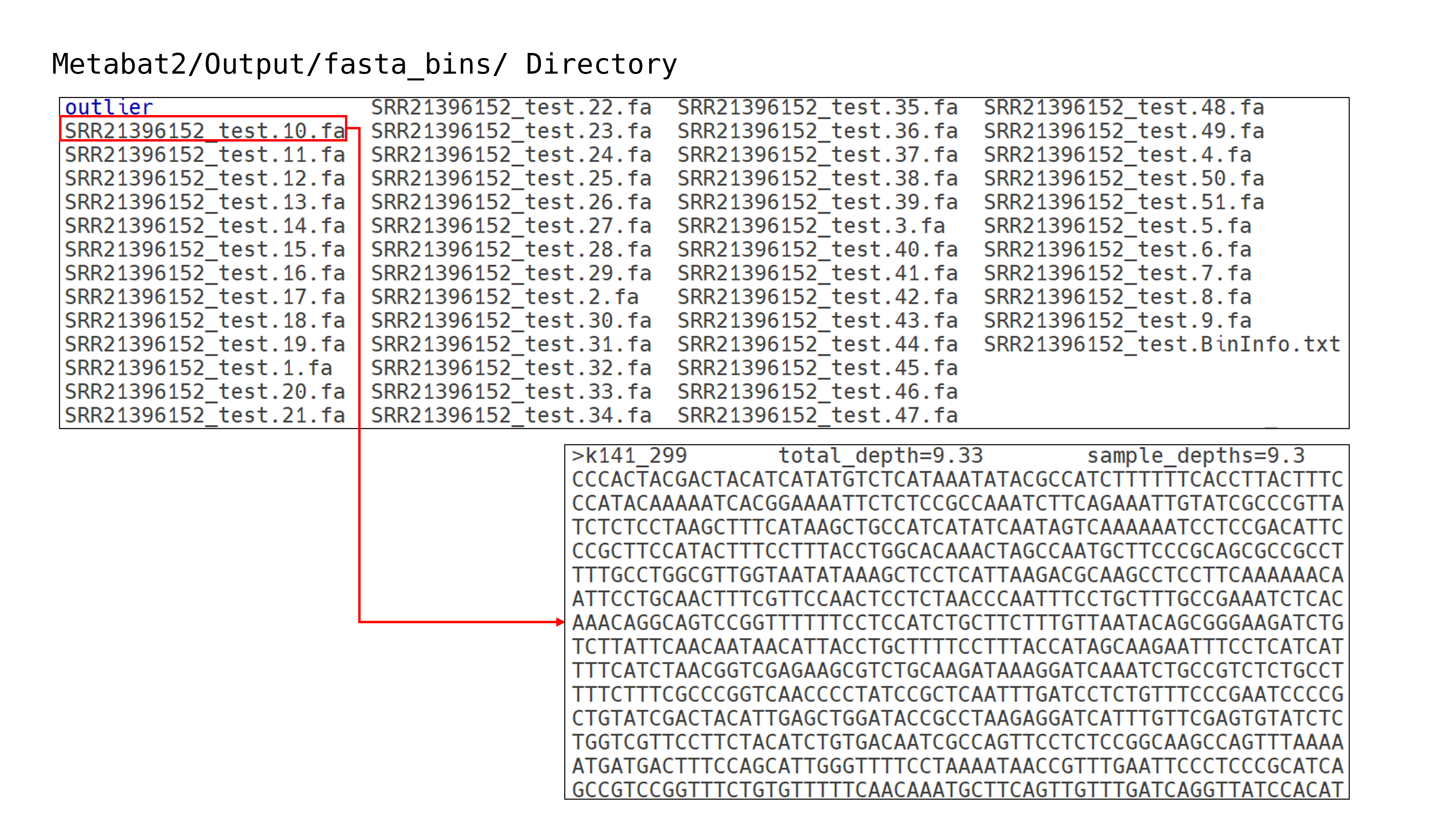
Metabat2/ouput/fasta_bins 디렉토리의 fasta 파일과 해당 파일의 구조. 이 디렉토리는 Metabat2 분석 결과 생성된 genome bin에 대한 FASTA 파일들이 위치해 있음. 각 파일에는 해당 bin에 소속된 contig 서열 정보가 담겨 있음.
-
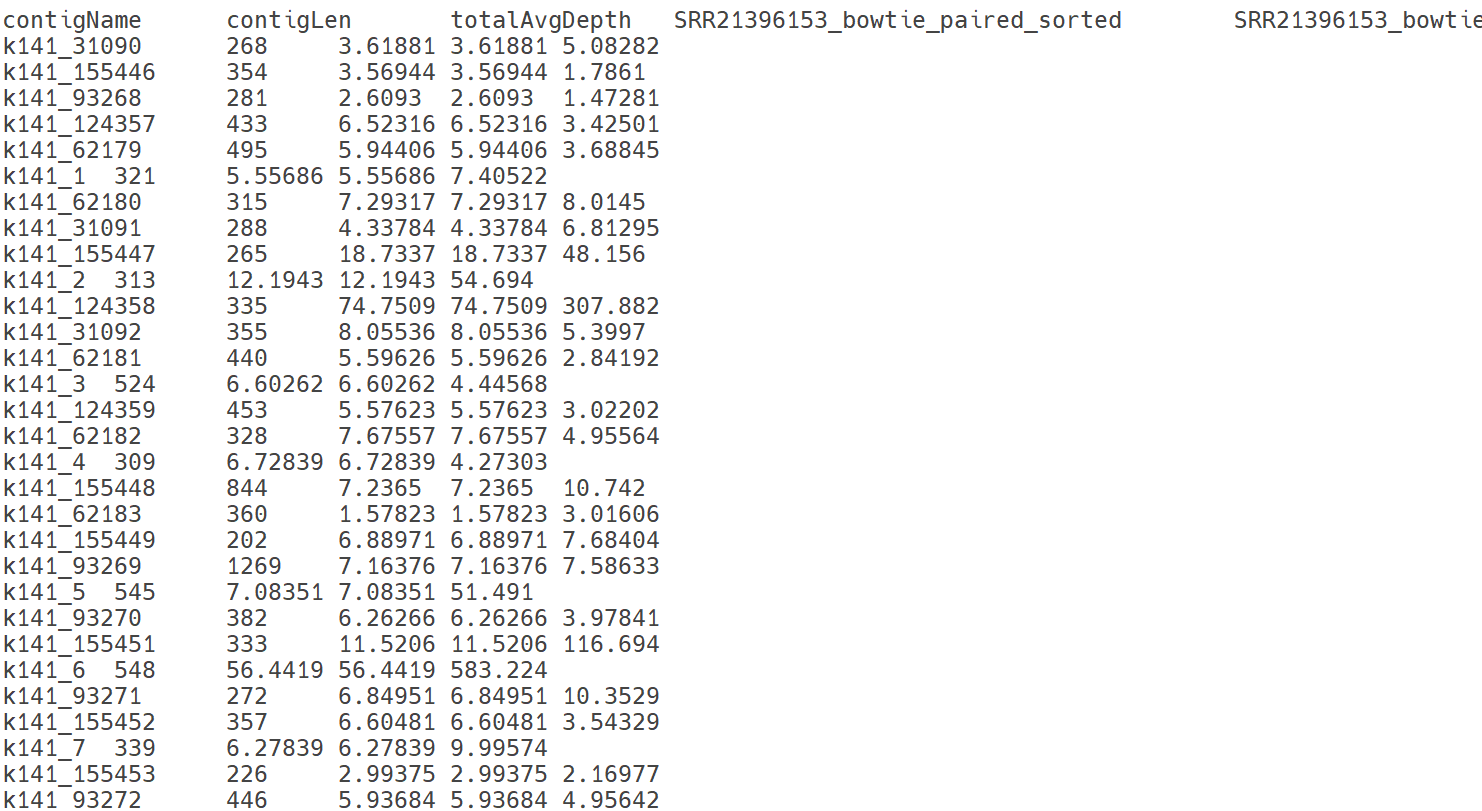
metabat2_depth 파일 내용. 각 contig의 시퀀싱 커버리지 수치에 대한 표로, 동일한 샘플에서 유래한 contig는 유사한 커버리지 수치를 가진다는 사실을 기반으로 Binning 과정에서 핵심 수치로 사용됨.
maxbin2
MaxBin2(Maximum-likelihood-based Binning 2)는 메타게놈 어셈블리 결과로부터 생성된 contig들을 염기서열 조성(tetranucleotide frequency; TNF), 커버리지 정보, 그리고 단일복제 마커 유전자(single-copy marker genes; SCGs)를 활용하여 자동으로 binning하는 도구입니다. Expectation-Maximization(EM) 알고리즘 기반의 확률 모델을 통해 각 contig이 특정 bin에 속할 가능성을 추정하며, SCGs를 이용해 bin의 개수 및 품질(완전도, 오염도)을 평가합니다. 이를 통해 혼합된 미생물 군집으로부터 개별 미생물 유전체(MAG)를 재구성할 수 있습니다.
주요사항
- MaxBin2는 정렬된 BAM 파일로부터 samtools depth를 이용해 contig별 커버리지를 계산하고 생성된 abundance_list를 입력으로 하여, binning을 수행함
- 본 프로그램은 후속 통합 분석에 사용할 수 있도록 DAS Tool의 입력 요구사항에 맞추어 결과 파일을 변환함
실행 명령어 예시
Maxbin2.sh \ input_dir = ./SAMtools_sort/output \ fasta_dir = ./megahit/output \ output_dir = ./maxbin2/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./SAMtools_sort/output | 분석에 사용될 정렬된 BAM 파일이 있는 디렉터리 경로 | |
| Input | Folder | fasta_dir | ./megahit/output | 석에 사용될 assembled fasta 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./maxbin2/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
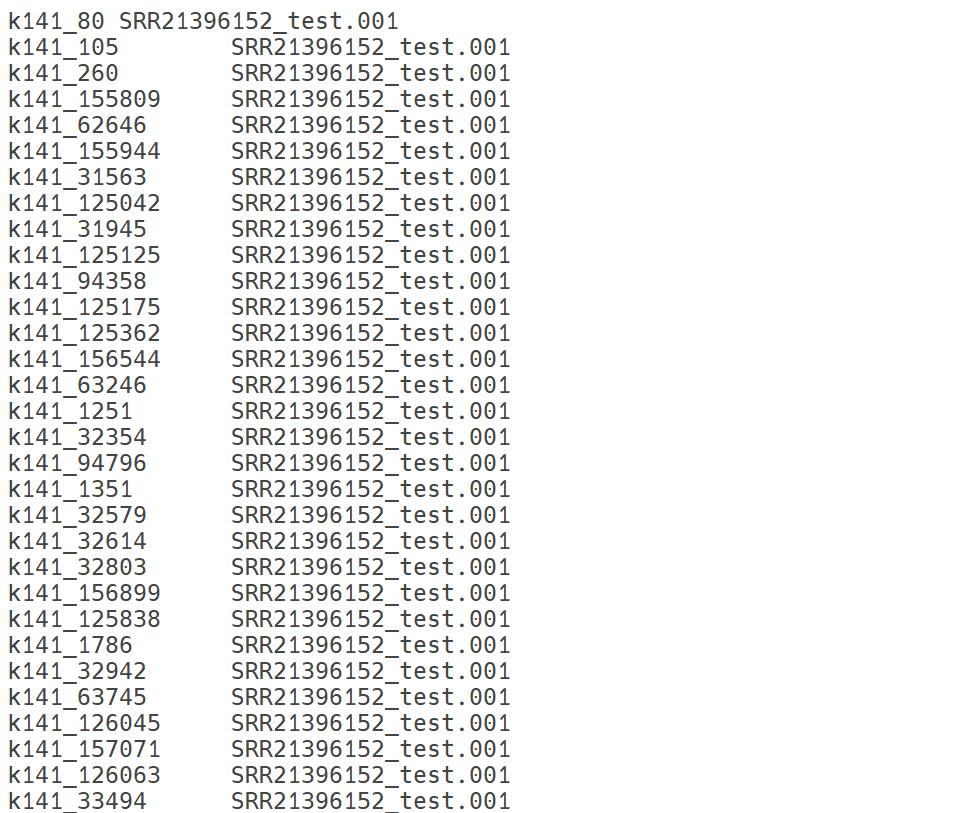
maxbin2_contigs2bin.tsv 파일 내용. Maxbin2 분석 결과 각 conitg와 그에 부여된 최종 genome bin ID를 매핑한 파일.
-
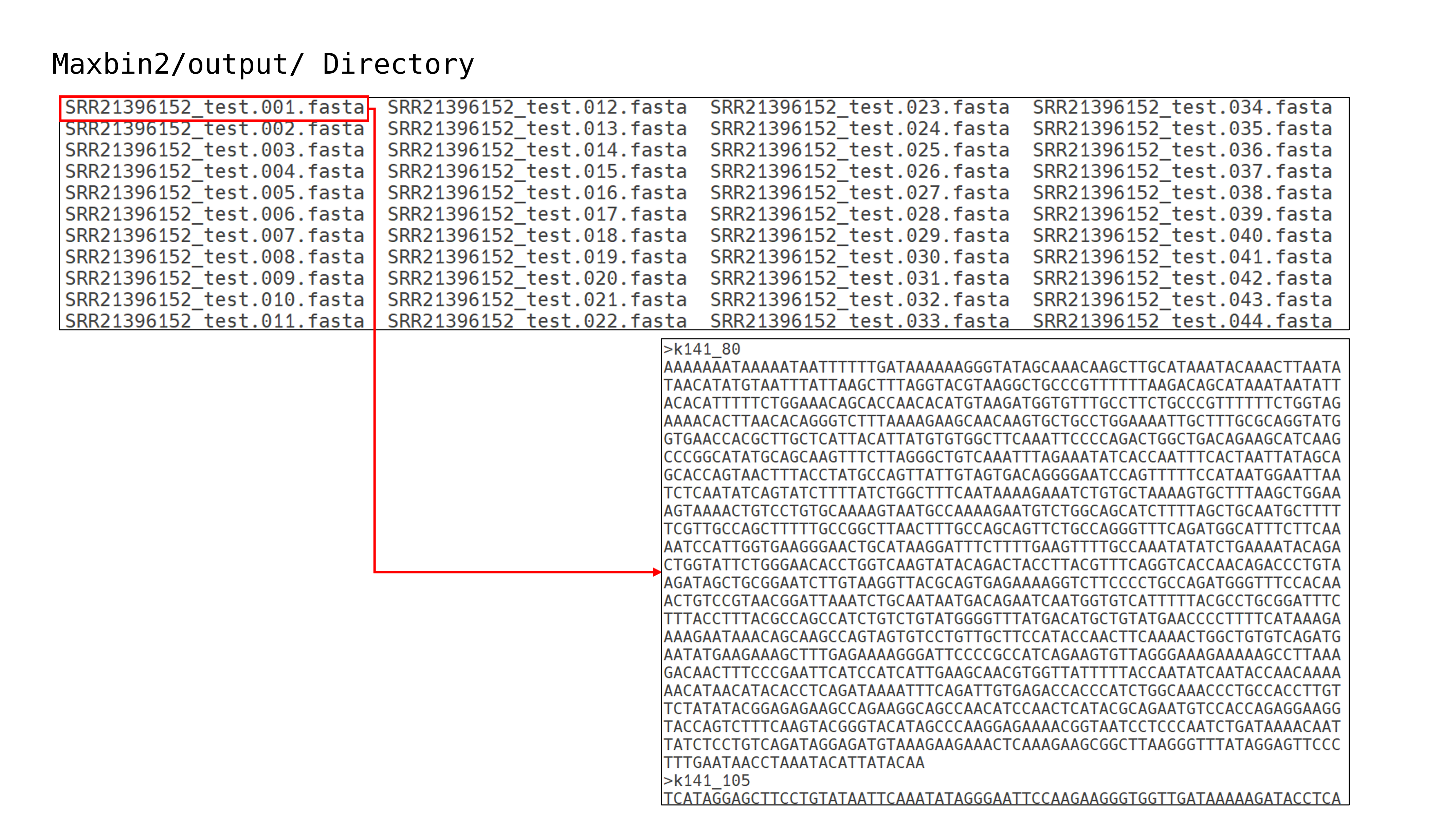
maxbin2/ouput/ 디렉토리의 fasta 파일과 해당 파일의 구조. 이 디렉토리는 Maxbin2 분석 결과 생성된 genome bin에 대한 FASTA 파일들이 위치해 있음. 각 파일에는 해당 bin에 소속된 contig 서열 정보가 담겨 있음.
-
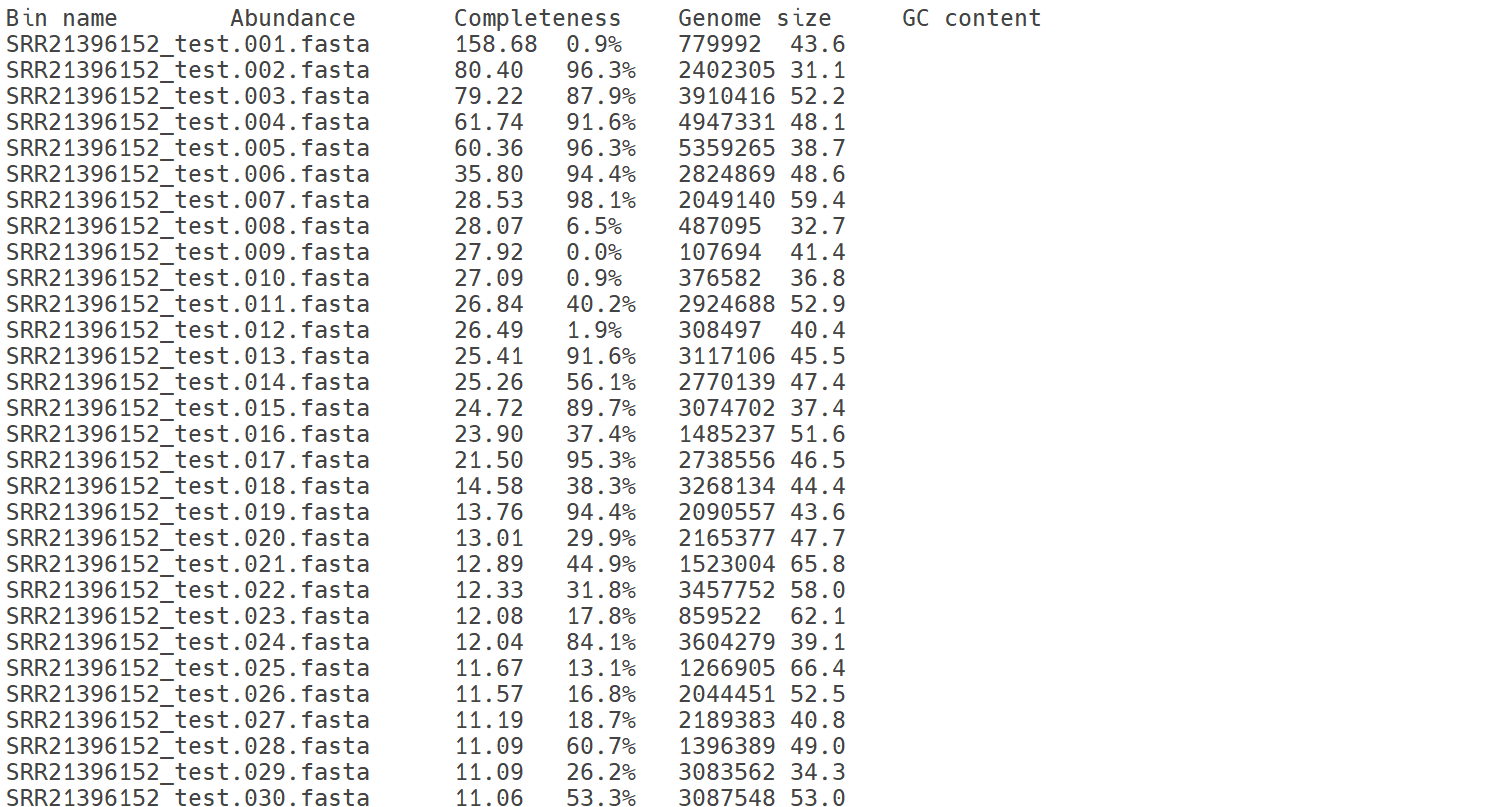
.summary 파일 내용. 각 genome bin에 대한 정보를 요약하고 있음. bin의 이름, 포함된 contig 수, 마커 유전자로 추정한 bin의 완성도 등에 대한 정보를 포함함.
-

.abund 파일 내용. 각 contig의 풍부도를 매핑한 표로, 여기서 풍부도는 시퀀싱 커버리지 수치와 같은 값임. 동일한 샘플에서 유래한 contig는 유사한 커버리지 수치를 가진다는 사실을 기반으로 binning 과정에서 핵심 수치로 사용됨.
-
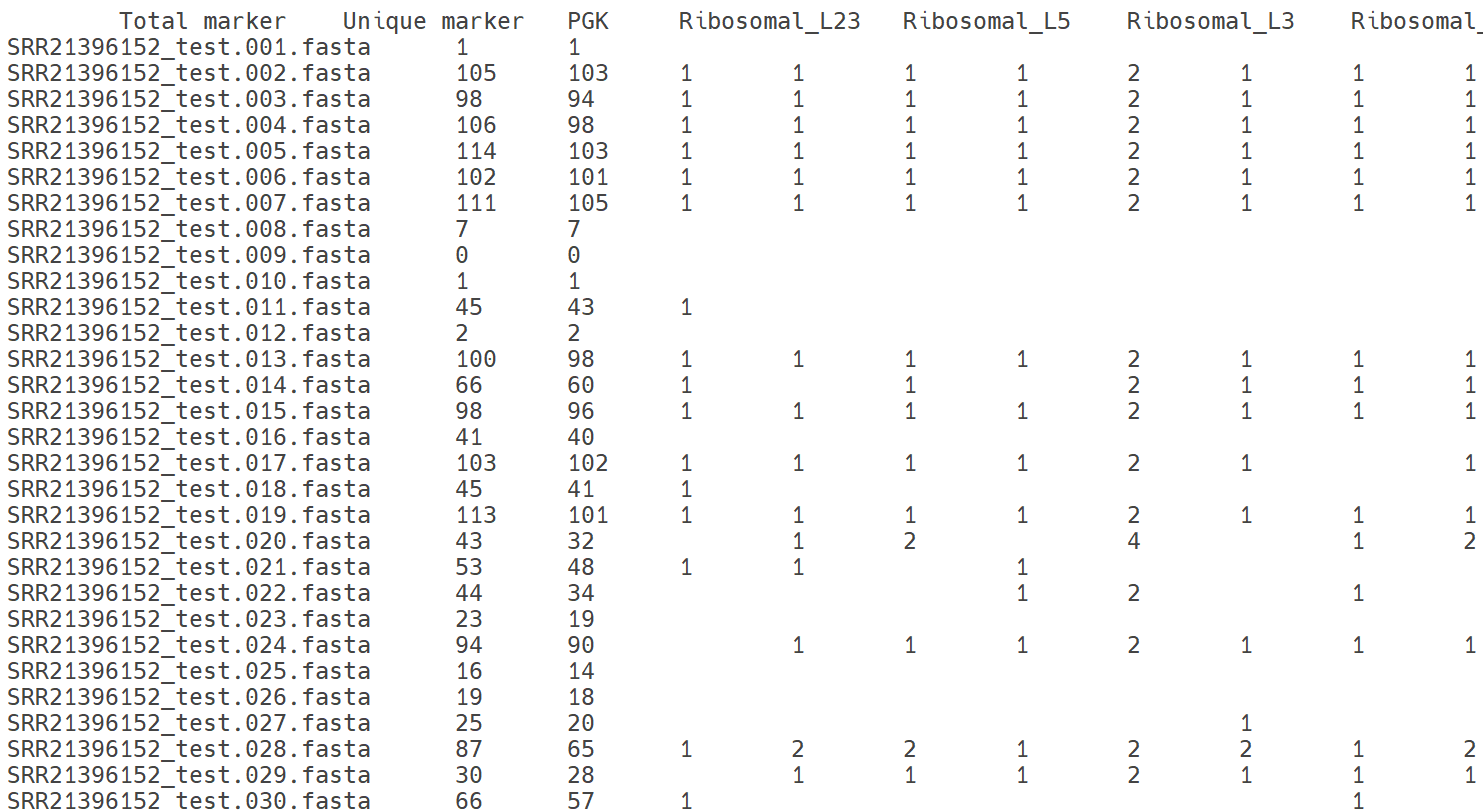
.marker 파일 내용. binning 과정에서 사용한 마커 유전자에 대한 정보를 담고 있음.
DAStool
DAS Tool은 여러 binning 도구(CONCOCT, MetaBAT2, MaxBin2 등)의 결과를 통합하여 각 bin의 품질을 개선하고 중복을 제거하기 위한 통합 binning 정제(refinement) 도구입니다. 개별 binning 결과로부터 얻은 bin들을 단일 세트로 병합한 후, 각 contig의 속성과 bin 품질 지표(완전도, 오염도, SCG 존재 여부 등)를 종합적으로 평가하여 점수화(scoring)합니다. 이후 점수가 가장 높은 bin 조합을 선택함으로써, 중복된 bin을 제거하고 완전도와 정확도가 향상된 최적의 MAG 세트를 생성합니다.
실행 명령어 예시
DAStool.sh \ input_cococt_dir= ./Concoct/output \ input_metabat2_dir= ./Metabat2/output \ input_maxbin2_dir= ./Maxbin2/output \ fasta_dir= ./megahit/output \ output_dir=./DAStool/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_cococt_dir | ./Concoct/output | 분석에 사용될 binning 결과 파일이 포함된 디렉터리 경로 | |
| Input | Folder | input_metabat2_dir | ./Metabat2/output | 분석에 사용될 binning 결과 파일이 포함된 디렉터리 경로 | |
| Input | Folder | input_maxbin2_dir | ./Maxbin2/output | 분석에 사용될 binning 결과 파일이 포함된 디렉터리 경로 | |
| Input | Folder | fasta_dir | ./megahit/output | 분석에 사용될 assembled fasta 파일이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./DAStool/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
.png)
_DASTool_contig2bin.tsv 파일 내용. DASTool 분석 결과 통합된 최종 genome bin에 대해 각각 그에 소속되는 contig와 bin ID를 매핑한 파일.
-
.png)
_DASTool_bins/ 디렉토리의 파일과 해당 파일의 구조. 이 디렉토리는 여러 binning 결과를 DASTool을 통해 최적화 및 통합한 최종 genome bin의 FASTA 파일들이 위치해 있음. 각 파일에는 해당 bin에 소속된 contig 서열 정보가 담겨 있음.
-
.png)
_DASTool_summary.tsv 파일 내용. 최종 genome bin에 대한 각종 수치를 담고 있음.
-
.png)
_proteins.faa 파일 내용. 각 contig 서열을 단백질 아미노산 서열로 번역한 정보가 담은 FASTA 형식의 파일.
-
.png)
_proteins.faa.bacteria.scg 파일 내용. DASTool은 bin의 완성도를 평가하기 위해 SCG(Single-Copy Gene)을 이용하는데, faa 파일에서 박테리아에 대한 SCG를 찾은 결과임. 고세균에 대한 결과 파일도 있음.
quast_dastool
QUAST(Genome Assembly Quality Assessment Tool)는 유전체 조립 결과(assembly)의 품질을 정량적으로 평가하기 위한 표준 도구입니다. 입력된 FASTA 서열(contig/scaffold)을 기반으로 contig 수, GC 비율, N50, L50, 최대 contig 길이 등 다양한 구조적 통계를 계산합니다. Reference genome을 함께 제공하면 alignment 기반의 정확도 지표(Genome fraction, Mismatch rate, Misassembly 수 등)도 산출할 수 있다. 일반적으로 단일 생물종의 WGS나 isolate genome 분석에 사용되며, DAS Tool 이후의 MAG bin 파일처럼 reference가 불명확한 경우에는 reference-free 모드로 사용해야 합니다. 결과물은 report.txt, report.tsv, report.html 형태로 제공됩니다.
주요사항
- QUAST는 단일 또는 다중 어셈블리 비교 가능함
- QUAST는 참조 서열 제공 시 오조립 탐지 및 정확도 평가를 수행할 수 있으며, 참조 서열을 제공하지 않는 경우에는 구조적 통계를 기반으로 QC를 수행함
실행 명령어 예시
quast_dastool.sh \ input_dir=./DASTool/output \ output_dir=./quast_dastool/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./DASTool/output | 분석에 사용될 binned fasta 파일들이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./quast_dastool/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 | |
| Option | Integer | min_contig | 500 | 결과 보고서에 포함할 최소 컨티그(contig) 길이 (default: 500) |
결과
-
.png)
output/ 디렉토리에 최종 선별된 genome bin 별로 개별 디렉토리가 생성됨. 각 디렉토리에는 해당 게놈에 대해 QUAST로 수행한 Assembly QC 결과가 담겨 있음.
-
.png)
report.txt 파일 내용. 입력 데이터의 contig 수, GC 함량 등 기본적인 수치와 assmebly의 완성도를 평가할 N50, L50 등의 품질 수치를 담고 있음.
-
.png)
report.pdf 파일 내용. report.txt의 수치 및 이를 시각화한 그래프를 담고 있음.
-
.png)
report.html 파일 내용. report.txt의 통계 수치 및 이를 시각화한 그래프, 그리고 contig 구조에 대한 genome map을 제공하고 있음.
checkM2
CheckM2는 유전체 또는 메타게놈 어셈블리 결과의 품질을 평가하기 위한 도구로, 단일 균주 유전체부터 메타게놈에서 추출된 MAG(Metagenome-Assembled Genome)에 이르기까지 폭넓게 적용할 수 있습니다. 인공지능(AI) 기반의 예측 모델을 활용하여 각 유전체의 완전도(completeness) 와 오염도(contamination)를 정량적으로 산출하며, 기존 CheckM이 단일복제 마커 유전자 기반의 규칙형(rule-based) 접근 방식을 사용했던 것과 달리, CheckM2는 대규모 reference 유전체 및 단백질 프로파일로 학습된 신경망 모델(Neural Network Model) 을 통해 정확도와 처리 속도를 크게 향상시켰습니다. 입력으로 FASTA 또는 단백질 서열 파일을 사용하며, 내부적으로 유전자 예측과 단백질 도메인 분석을 수행한 후 품질 지표를 계산합니다. 이를 통해 개별 유전체 및 메타게놈 bin(MAG)의 신뢰성과 품질을 효과적으로 평가할 수 있습니다.
주요사항
- CheckM2는 UniRef100 단백질 서열 기반의 CheckM2 reference database를 사용함 (예: uniref100.KO.1.dmnd). 해당 DB는 AI 모델 학습 및 품질 예측 시 필수로 사용되며, 버전 불일치 시 결과 재현성에 영향 가능
실행 명령어 예시
checkm2.sh \ input_dir=./DAStool/output \ output_dir=./checkm2/output \
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./DAStool/output | 분석에 사용될 binned fasta 파일들이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./checkm2/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
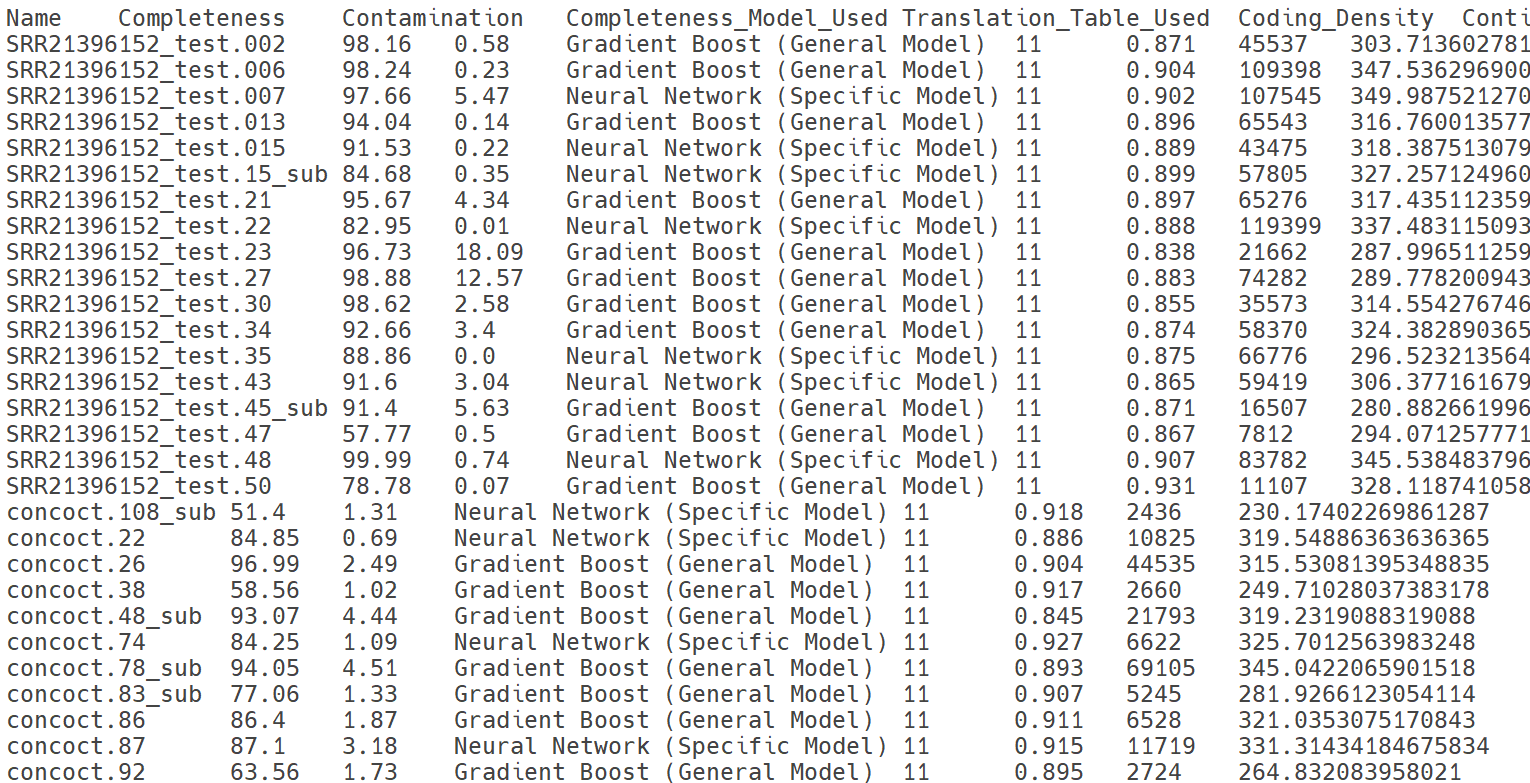
quality_report.tsv 파일 내용. CheckM2 분석 결과의 최종 요약 리포트. 각 행은 하나의 genome bin을 나타냄. bin ID, 예측된 bin의 완성도(Completeness), 예측된 오염도(Contamination) 등의 핵심 품질 수치를 담고 있음.
prokka
Prokka(Rapid Prokaryotic Genome Annotation)는 세균 및 고세균 유전체를 빠르고 표준화된 방식으로 주석(annotation)하기 위한 도구입니다. FASTA 형식의 유전체 서열을 입력으로 받아, CDS(단백질 코딩 서열), tRNA, rRNA, 기타 기능적 유전자 영역을 자동 탐지하고, 내부의 단백질 데이터베이스(RefSeq, UniProt, HAMAP 등)와 비교하여 유전자 기능을 부여합니다. 다중 스레드 기반으로 빠르게 수행되며, 표준화된 GFF, GBK, FAA, FNA 등의 포맷으로 결과를 출력합니다. 이를 통해 MAG을 포함한 다양한 미생물 유전체에 대해 신속하고 일관된 주석 결과를 제공할 수 있습니다.
실행 명령어 예시
prokka.sh \ input_dir=./DAStool/output \ output_dir=./prokka/output \
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./DAStool/output | 분석에 사용될 binned fasta 파일들이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./prokka/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-
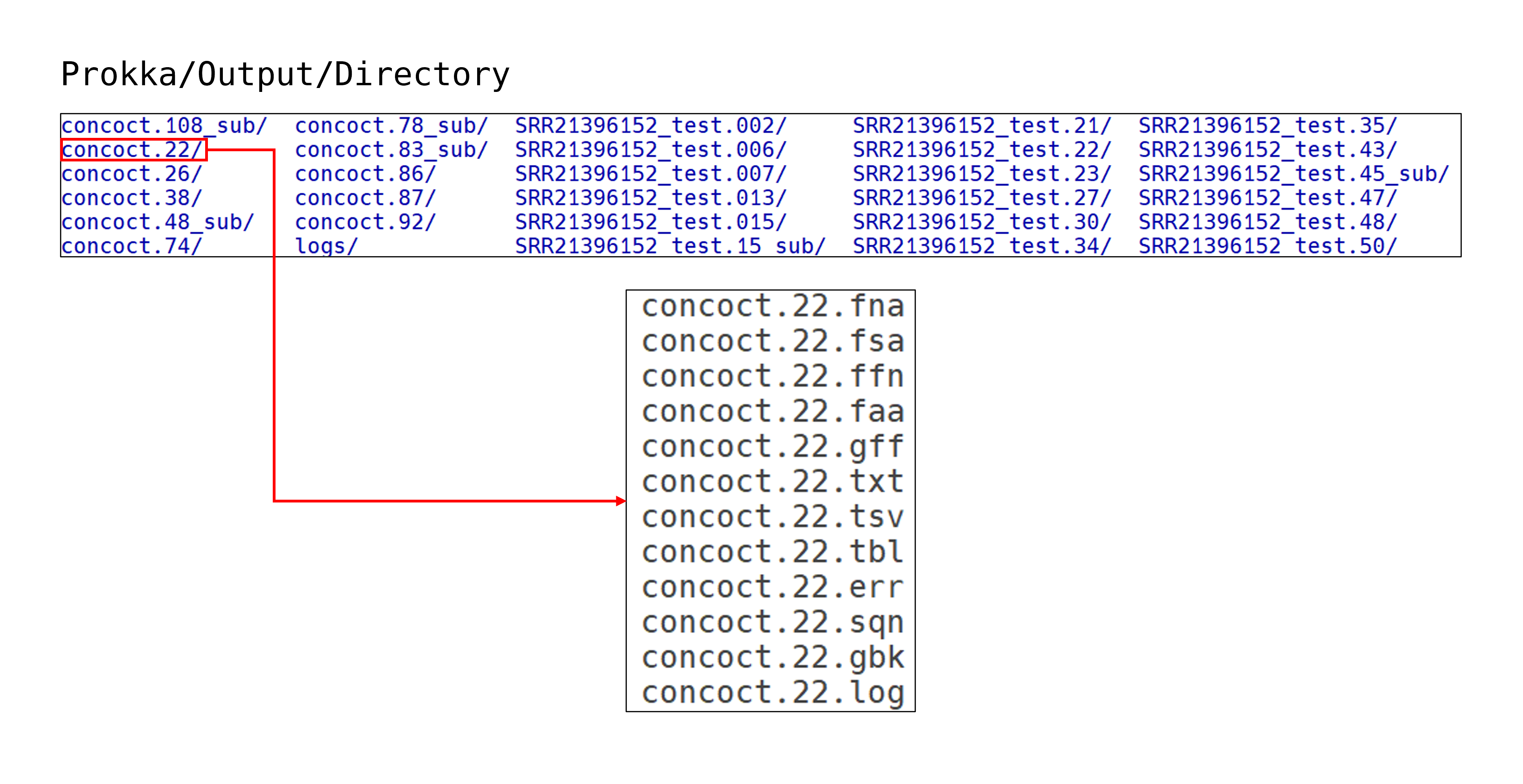
output/ 디렉토리에 최종 선별된 genome bin 별 개별 디렉토리가 생성됨. 각 폴더는 해당 게놈 서열에 대해 Prokka로 수행한 유전자 주석 결과를 담고 있음. 주요 파일 포맷으로 gbk, gff, faa 등이 있음.
-
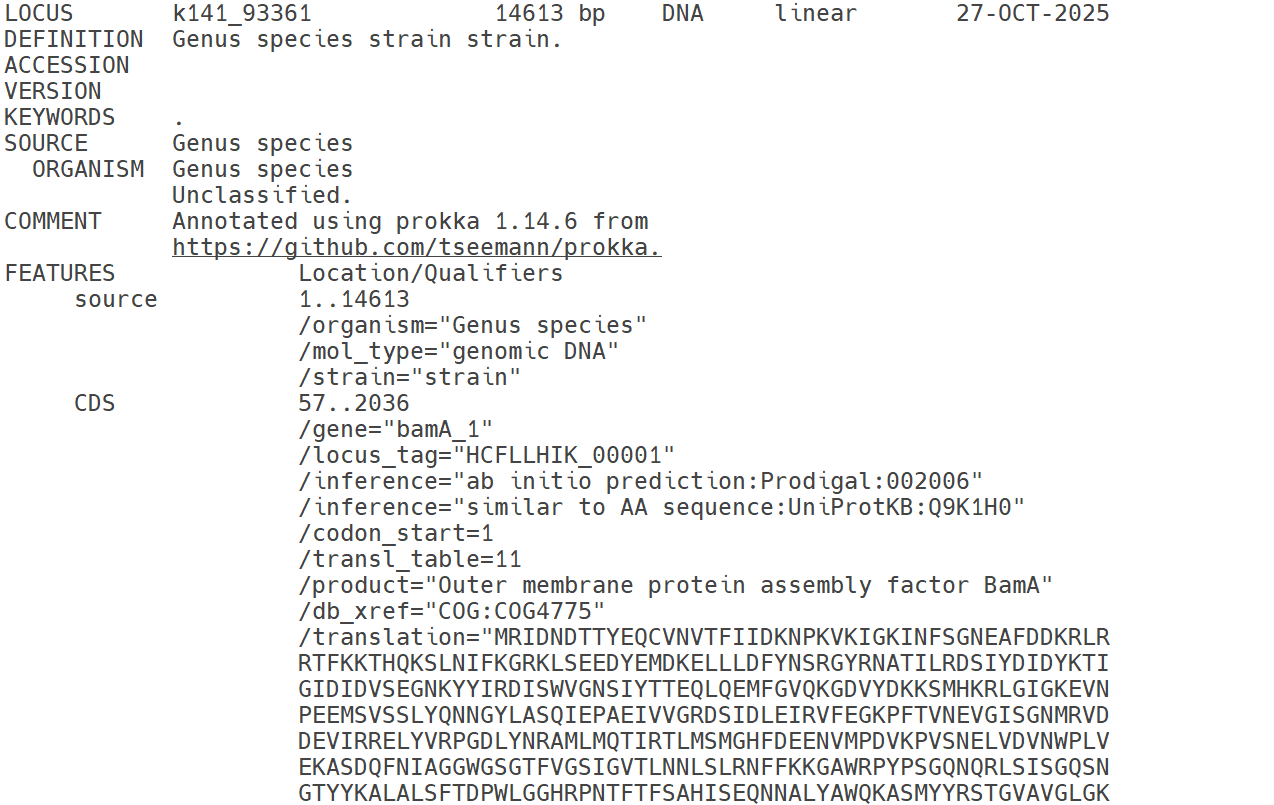
gbk 파일 내용. .gbk 파일 내용. GenBank 포맷은 NCBI에서 만든 주석 파일 형식으로, 예측된 유전자, rRNA, tRNA 등의 위치 및 기능 정보가 포함됨. LOCUS, DEFINITION, SOURCE 등 서열 정보가 있는 header와 주석 정보가 담긴 feature 부분으로 구성됨.
-
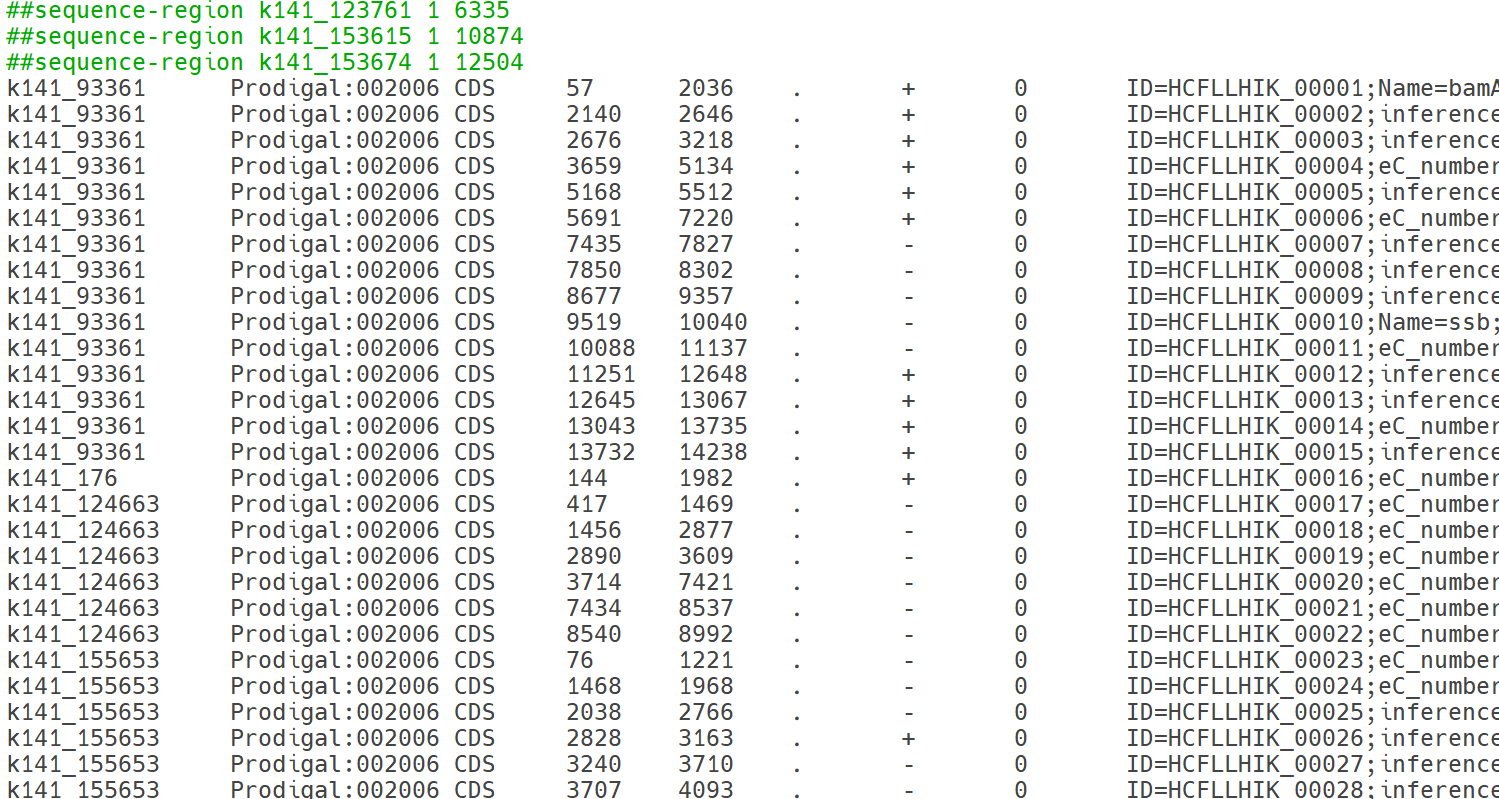
gff 파일 내용. 대표적인 주석 파일 형식으로, 예측된 각 유전자에 대해 서열 이름, 시작점, 끝점, 방향과 같은 위치 정보와 score, start codon type 등의 상세 정보가 포함됨.
-

faa 파일 내용. 예측된 각 유전자 서열을 단백질 아미노산 서열로 번역한 정보를 담은 FASTA 형식의 파일.
GTDB-Tk
GTDB-Tk(Genome Taxonomy Database Toolkit)는 유전체의 계통 분류(taxonomic classification)를 수행하기 위한 도구로입니다. 완전한 유전체 어셈블리부터 메타게놈으로부터 추출된 MAG(Metagenome-Assembled Genome) 데이터까지 폭넓게 적용할 수 있습니다. GTDB에 포함된 표준화된 계통 분류 체계를 기반으로, 각 유전체의 유전적 거리와 단일복제 마커 유전자의 계통적 위치를 분석하여 분류 계층(계–문–강–목–과–속–종)을 할당합니다. 내부적으로 marker gene alignment, phylogenetic placement, classification 단계를 자동으로 수행하며, RAxML, FastANI 등의 외부 도구를 통합하여 정확하고 재현성 있는 계통 분류를 제공합니다. 이를 통해 다양한 유전체 데이터에 대해 일관되고 재현성 있는 계통 정보를 부여할 수 있습니다.
주요사항
- 이 프로그램은 Genome Taxonomy Database release 226 (R226) 을 기반으로 동작하며, 해당 DB에는 표준화된 세균 및 고세균 참조 유전체가 포함되어 있음.
실행 명령어 예시
GTDB_Tk.sh \ input_dir=./input \ output_dir=./GTDB_Tk/output
실행 스크립트
파라미터
| 옵션 | 유형 | 명칭 | 값 | 설명 | 필수값 |
|---|---|---|---|---|---|
| Input | Folder | input_dir | ./input | 분석에 사용될 binned fasta 파일들이 있는 디렉터리 경로 | |
| Output | Folder | output_dir | ./gtdb_tk/output | 분석 실행 후 결과 파일이 저장될 디텍터리 경로 |
결과
-

.bac120.summary.tsv 파일 내용. 입력한 genome bin의 최종 계통 분류 결과. 계통 분류에 사용된 GTDB 내 참조 게놈과 ANI 값 및 각종 수치를 담고 있음.